



CorSalud 2015 Jul-Sep;7(3):229-234
CASO CLÍNICO
DISPLASIA ARRITMOGÉNICA DEL VENTRÍCULO DERECHO. PRESENTACIÓN DE UN CASO
Dra. Tessa Negrín Valdés, Dra. Livian M. Lage López, Dr. Guillermo R. Quintana Cañizares, Dr. Alexander Santos Pérez y Dra. Amarilys Valero Hernández
______________
Servicio de Cardiología. Hospital General Universitario Camilo Cienfuegos. Sancti Spíritus, Cuba.
Correspondencia: T Negrín Valdés. Hospital Camilo Cienfuegos. Bartolomé Masó N° 128. Sancti Spíritus, CP 60100. Sancti Spíritus, Cuba. Correo electrónico: tessa@hpss.ssp.sld.cu
Resumen
La displasia arritmogénica de ventrículo derecho es una enfermedad del músculo cardíaco que afecta predominantemente al mencionado ventrículo, provoca el reemplazo del miocardio normal por tejido adiposo o fibroadiposo y es causa de muerte súbita en individuos jóvenes. La manifestación clínica relevante es la taquicardia ventricular, aunque se han descrito casos de insuficiencia cardíaca derecha o global. El diagnóstico se confirma con ecocardiografía y resonancia magnética nuclear. En este artículo se presenta a un paciente exfumador, de 65 años de edad, con hipertensión arterial sistémica y cardiopatía isquémica, con antecedentes de cuadros sincopales al esfuerzo y presencia demostrada de taquicardias ventriculares no sostenidas, con morfología de bloqueo de rama izquierda. Se realizaron los estudios diagnósticos pertinentes y se constataron elementos ecocardiográficos compatibles con displasia arritmogénica de ventrículo derecho, por lo que se implantó un desfibrilador automático implantable, tras lo cual el paciente ha evolucionado favorablemente.
Palabras clave: Displasia arritmogénica , Ventrículo derecho, Arritmia, Bloqueo de rama izquierda
Arrhythmogenic right ventricular dysplasia: A case report
Abstract
Arrhythmogenic right ventricular dysplasia is a heart muscle disease that predominantly affects the right ventricle, bringing about the replacement of normal myocardium with fatty or fibrofatty tissue and causing sudden death in young individuals. Ventricular tachycardia is an important clinical manifestation, although there are reports of right or global heart failure. The diagnosis is confirmed by echocardiography and magnetic resonance imaging. The case of a 65-year-old former smoker, with hypertension and ischemic heart disease, a history of effort syncope symptoms and proven non-sustained ventricular tachycardia, with morphology of left bundle branch block, is reported. Relevant diagnostic studies were performed, and echocardiographic elements which were compatible with arrhythmogenic right ventricular dysplasia were found. Therefore, an implantable cardioverter defibrillator was implanted, after which the patient has had a favorable outcome.
Key words: Arrhythmogenic dysplasia, Right ventricle, Arrhythmia, Left bundle branch block
Introducción
La displasia arritmogénica de ventrículo derecho (DAVD) ha sido denominada como una miocardiopatía arritmogénica caracterizada por una pérdida progresiva de los miocardiocitos por reemplazo de tejido fibroso y adiposo, asociado al incremento del riesgo de arritmias y muerte súbita1. Se plantea que su causa genética está vinculada a una herencia autosómica dominante en un tercio de los casos, el rasgo recesivo posee una alta penetrancia. El ventrículo derecho (VD) es predominantemente afectado, involucrado en un 60 % de los casos2. Sin embargo, en estadios avanzados de la enfermedad puede desarrollarse una insuficiencia cardíaca biventricular.
El diagnóstico y la diferenciación de la DAVD es un verdadero desafío. Los criterios clínicos para su identificación tienen una baja sensibilidad en estadios tempranos. Electrocardiográficamente se caracteriza por la presencia de ondas épsilon y morfología de bloqueo de rama derecha. La ventriculografía de contraste o la biopsia endomiocárdica se utilizan cuando los métodos incruentos no son concluyentes3.
Las variantes terapéuticas incluyen la terapia con fármacos antiarrítmicos, guiada por estimulación ventricular programada; así como la ablación por radiofrecuencia y la cirugía. El desfibrilador automático implantable (DAI), solo o en combinación con el tratamiento farmacológico, tiene una importancia relevante4.
El caso que se presenta constituyó un reto para el diagnóstico, por no contar con la confirmación genética y realizarse a través de medios incruentos. Su evolución posterior fue favorable.
Caso clínico
Paciente exfumador, de 65 años de edad, antecedentes de hipertensión arterial sistémica y cardiopatía isquémica, y varios ingresos por síncope de esfuerzo sin diagnóstico definitivo. En el último episodio se observó un bloqueo de rama izquierda del haz de His (BRIHH), que se interpretó como agudo y de probable causa isquémica (Figura 1), por lo que fue ingresado en la Unidad de Cuidados Intensivos Coronarios bajo monitorización estricta, y se comenzaron estudios por esta causa.
En las primeras 24 horas posteriores al ingreso se pudieron constatar taquicardias ventriculares (TV) no sostenidas con morfología de BRIHH durante el síncope (Figura 2).
Se decidió iniciar tratamiento de impregnación oral con amiodarona 400 mg/día, asociado a tratamiento antiisquémico con antiagregación oral, nitroglicerina intravenosa a 0,5 mcg/kg/min y enalapril 20 mg/día.
Como parte del estudio se realizó ecocardiograma basal con hallazgos imagenológicos compatibles con DAVD (Figura 3):
– Septum interventricular 13 mm, pared posterior del ventrículo izquierdo (VI) 5 mm, diámetro diastólico del VI 66 mm, diámetro sistólico del VI 56 mm, volumen diastólico final 226 ml, volumen sistólico final 179 ml, fracción de eyección del VI 20,9 % por el método de Teichholz y 22,5 % por Simpson.
– Diámetro diastólico medio del VD 46 mm, con disquinesia a nivel de los segmentos apicales e inferobasales. Incremento del grosor de la banda moderadora con aspecto trabeculado, dilatación irregular del tracto de salida del VD de 39mm, excursión sistólica de la valva anterior de la tricúspide de 9 mm. Función sistólica del VD gravemente deprimida.
El paciente evolucionó sin nuevos episodios sincopales, con estabilidad clínica y hemodinámica, y fue remitido al centro de referencia territorial (Cardiocentro Ernesto Che Guevara), con el fin de definir la causa del cuadro clínico. En la coronariografía se encontró una arteria coronaria derecha con calcificaciones crónicas y difusas, en segmentos medio y distal, con buenos lechos distales, sin criterio de intervencionismo coronario percutáneo por ausencia de lesiones significativas.
Se coordinó con el Servicio de Arritmias y Electrofisiología del Instituto de Cardiología y Cirugía Cardiovascular de La Habana (centro de referencia nacional) y se complementó el estudio, donde se corroboró el diagnóstico presuntivo y se implantó un DAI como tratamiento definitivo.
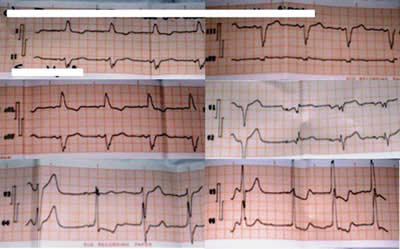
Figura 1. Electrocardiograma previo al episodio sincopal, realizado en la Unidad de Cuidados Intensivos Coronarios, que muestra un BRIHH.
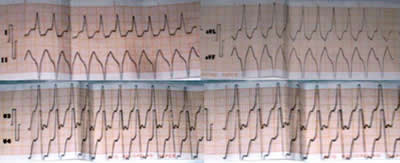
Figura 2. Electrocardiograma durante el síncope que muestra taquicardia con QRS ancho y disociación aurículo-ventricular compatible con TV no sostenida con morfología de BRIHH.
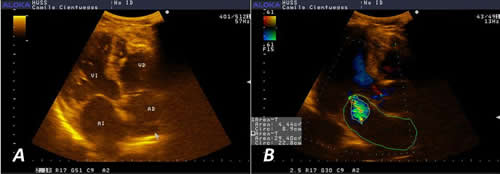
Figura 3. Ecocardiograma. A. Vista apical donde se observa la dilatación de todas las cavidades cardíacas y los cambios anatómicos del VD. B. Insuficiencia mitral leve.
Comentario
Las primeras referencias de la DAVD las realizó Dalla Volta en 1961, pero solo en 1977, tras la muerte de un joven médico italiano mientras practicaba tenis es que se inicia la investigación profunda de esta enfermedad5. Un año después, se hace la descripción definitiva por Fontaine et al.6.
Su prevalencia es estimada en 1:5000 en la población general, lo que indica que la muerte súbita puede ser el primer síntoma, sobre todo en personas jóvenes, especialmente atletas. No obstante, se han publicado series en pacientes ancianos.
Esta enfermedad se asocia a taquiarritmias ventriculares por reentrada con origen en el VD, por lo que muestra una morfología eléctrica de BRIHH. Pueden aparecer palpitaciones inducidas por el ejercicio, fatiga, mareos, dolor torácico atípico y en algunos casos, síncope. Un gran número de pacientes son asintomáticos y la primera manifestación es un episodio de muerte súbita que ocurre frecuentemente mientras se realiza actividad física. Las arritmias ventriculares –desde extrasístoles aislados hasta TV con patrón electrocardiográfico de BRIHH–, son las formas características de presentación7.
El patrón de herencia es autosómica dominante en un tercio de los casos y el rasgo recesivo posee una alta penetrancia, localizándose la anomalía genética en los cromosomas 1 y 14q23 y q24, y más recientemente se identificó el cromosoma 10 con aproximadamente 12 genes y locus vinculados a esta enfermedad.
Estas alteraciones determinan codificaciones para proteínas desmosomales, identificadas como Plakoglobin (JUP), Desmoplakin (DSP), Plakophilin-2 (PKP2), Desmoglein-2 (DSG2) y Desmocollin-2 (DSC2). La adhesión célula-célula depende ampliamente de la interacción intracelular desmosomal especialmente de DSP y JUP. La deficiencia de DSP provoca un incremento en la expresión de los genes adipogénicos y fibrogénicos, y el depósito anormal de tejido fibro–adiposo, por esta razón, también se ha denominado como una enfermedad unional célula–célula8.
Desde el punto de vista anatomopatológico se observa adelgazamiento de la pared ventricular e infiltración grasa de las áreas subepicárdicas. Se puede mostrar un VD dilatado con protrusiones en las zonas infundibular, apical y subtricuspídea, en el llamado triángulo de la displasia. En la microscopía se observa el reemplazo de las capas externa y media del miocardio ventricular derecho y, en menor extensión del izquierdo, por tejido adiposo y fibrosis que limitan tiras o grupos de fibras miocárdicas. Existen zonas de fibras normales o miofribrillas parcialmente degeneradas que son las responsables de la conducción lenta y del fenómeno de reentrada, origen de las TV9.
El grupo de trabajo en enfermedades miocárdicas y pericárdicas de la Sociedad Europea de Cardiología y el Consejo Científico en Miocardiopatías han propuesto algunos criterios para facilitar el diagnóstico de esta enfermedad, con diferentes formas de presentación clínica, pero tienen una baja sensibilidad en estadios tempranos de la enfermedad10.
El hallazgo electrocardiográfico establecido como criterio mayor es la presencia de ondas épsilon, observadas en un 30 % de los casos, ubicadas en el comienzo del segmento ST en derivaciones precordiales derechas, por presencia de potenciales tardíos o la prolongación del QRS mayor de 110 ms de V1-V3.
Se plantean criterios menores como: ondas T invertidas de V2-V6 en mayores de 12 años, sin bloqueo de rama derecha del haz de His; TV sostenida o no con patrón de BRIHH, y extrasistolia ventricular frecuente (> 1.000 en 24 horas).
Entre los criterios morfofuncionales que pueden evaluarse por ecocardiografía y resonancia magnética nuclear (RMN) se encuentran como criterios mayores: la dilatación grave y la reducción de la fracción de eyección del VD, sin afectación (o afectación leve) del VI, así como aneurismas localizados del VD con dilatación segmentaria grave. Como criterios menores: dilatación leve global del VD o reducción de su fracción de eyección, o ambas, con VI normal; y dilatación segmentaria leve e hipoquinesia regional del VD. Se añaden otros criterios mayores importantes: hallazgo histopatológico de tejido fibroadiposo en sustitución del tejido miocárdico que puede evaluarse por RMN o biopsia endomiocárdica, y la historia familiar de DAVD diagnosticada. El antecedente familiar de muerte súbita en personas menores de 35 años con sospecha de la enfermedad, constituye un criterio menor.
De esta manera el diagnóstico se sustenta en la existencia de dos criterios mayores o de uno mayor con dos menores o, en su defecto, en la existencia de cuatro criterios menores.
Para llegar al diagnóstico del caso que se presenta se determinaron dos criterios ecocardiográficos mayores y uno electrocardiográfico definido como mayor, que lo constituyó el oportuno hallazgo de la TV no sostenida con morfología de BRIHH.
Actualmente se propone el empleo de otras técnicas combinadas de electrocardiografía de señal promediada con la RMN, de gran ayuda en la diferenciación de DAVD y TV derecha idiopática11. No obstante, en presencia de hallazgos ecocardiográficos típicos, la RMN y la angiografía pueden ser evitados. La tomografía axial muestra el compromiso localizado o difuso, la dilatación del VD, el afinamiento de su pared y su hipoquinesia. El hecho distintivo es el marcado incremento de la grasa subepicárdica delineado por análisis densitométrico de la imagen tomográfica. Se incluye además, el estudio electrofisiológico y la cineangiografía de VD12.
En la actualidad no existe una pauta establecida para el tratamiento; por ello, es individualizado dirigido esencialmente al control de arritmias ventriculares malignas. La farmacoterapia es de elección en pacientes con arritmias bien toleradas, que no impliquen riesgo para la vida. Si la función ventricular izquierda es normal, los fármacos antiarrítmicos de clase I pueden ser útiles. Si la función ventricular está deprimida, la amiodarona es el medicamento preferido por algunos autores y puede combinarse con betabloqueadores13.
Los pacientes con TV sostenida o fibrilación ventricular deben recibir terapia guiada por estimulación ventricular programada. El DAI es la más efectiva para prevenir la muerte súbita, debido a su capacidad de proveer terapia antitaquicardia y choque eléctrico. Representa la primera opción en pacientes con síncope o paro cardíaco y TV no inducible. Otra interesante medida terapéutica es la ablación por radiofrecuencia, aunque la recurrencia de TV se observa en alrededor de la mitad de los casos y estas recurrencias pueden provocar muerte súbita14.
La opción de aislar quirúrgicamente la pared ventricular derecha se utiliza en casos seleccionados, para prevenir la transmisión de ritmos anormales al VI, lo cual es discutido por la importancia que se la otorga en la actualidad a la disfunción del VD en las diversas cardiopatías. El trasplante cardíaco solo está indicado en casos de extrema dilatación ventricular derecha o incontrolable compromiso hemodinámico, aunque en la práctica es infrecuente esta forma de terapia para controlar arritmias cardíacas15.
El diagnóstico de esta enfermedad se torna difícil cuando no se dispone de todos los medios, pero el conocimiento y la actualización constante de sus detalles son muy relevantes para definir una conducta terapéutica adecuada que permita salvar la vida del paciente.
La existencia de sutiles datos clínicos y electrocardiográficos debe conducir a continuar investigando la presencia de la DAVD, con lo cual podría adquirir, probablemente una mayor prevalencia de la que se reconoce en la actualidad.
Recibido: 18 de septiembre de 2014
Aceptado: 4 de noviembre de 2014
Subir

















