



CorSalud 2014 Abr-Jun;6(2):181-192
ARTÍCULO DE REVISIÓN
PAPEL DEL ESTRÉS OXIDATIVO EN LA PATOGÉNESIS DE LA HIPERTENSIÓN ARTERIAL
Dr. Yosit Ponce Gutiérreza, Dr. Arik Ponce Gutiérreza, MSc. Dr. Arnaldo Rodríguez Leónb y Katherin Cabrera Garcíaa
______________
Policlínico "Juan B. Contreras Fowler". Ranchuelo, Villa Clara, Cuba.
Hospital Universitario "Dr. Celestino Hernández Robau". Santa Clara, Villa Clara, Cuba.
Correspondencia: Y Ponce Gutiérrez. Camilo Cienfuegos N° 63, e/ Carmen Rivero y Federico Escobar. Ranchuelo, Villa Clara, Cuba. Correo electrónico: froilanponce@capiro.vcl.sld.cu
Resumen
La producción aumentada de las especies reactivas de oxígeno ha sido implicada con varias enfermedades crónicas, incluida la hipertensión arterial. El estrés oxidativo es, a su vez, causa y consecuencia de esta hipertensión. La mayor fuente de especies reactivas de oxígeno cardiovascular, renal y neural es la enzima NADPH oxidasa. El estrés oxidativo se relaciona con disfunción endotelial, inflamación, hipertrofia, apoptosis, migración celular, fibrosis y angiogénesis; procesos importantes involucrados en la remodelación vascular de la hipertensión arterial. A pesar de la gran cantidad de datos que implican al estrés oxidativo como un factor causante de la hipertensión experimental, los resultados en humanos son menos conclusivos. El objetivo de esta revisión bibliográfica es describir el papel del estrés oxidativo en la fisiopatología de la hipertensión arterial. La mejor comprensión de estos mecanismos permitirá establecer una conducta más integral ante esta frecuente enfermedad.
Palabras clave: Estrés oxidativo, Especies reactivas del oxígeno, Hipertensión arterial
Role of oxidative stress in the pathogenesis of hypertension
Abstract
The increased production of reactive oxygen species has been involved in several chronic diseases, including hypertension. Oxidative stress is, in turn, cause and consequence of this hypertension. The enzyme NADPH oxidase is the major source of reactive species of cardiovascular, renal and neural oxygen. Oxidative stress is associated with endothelial dysfunction, inflammation, hypertrophy, apoptosis, cell migration, fibrosis and angiogenesis; important processes involved in vascular remodeling of hypertension. Despite the large amount of data that involve oxidative stress as a causative factor of experimental hypertension, results in humans are less conclusive. The aim of this review is to describe the role of oxidative stress in the pathophysiology of hypertension. A better understanding of these mechanisms will allow a more comprehensive behavior to this common disease.
Key words: Oxidative stress, Reactive oxygen species, Hypertension
Introducción
La hipertensión arterial (HTA) es una enfermedad crónica en que la tensión arterial (TA) se encuentra con cifras de 140/90 o más. En el mundo, cerca de un billón de personas padecen de HTA, y se estima que esta cifra puede aumentar a 1,5 billones para el año 2025. La génesis exacta es desconocida, pues solo aproximadamente el 5 % de los pacientes hipertensos presenta una causa precisa1.
A nivel molecular, numerosos factores han sido implicados en la fisiopatología de la HTA, como la activación del sistema renina-angiotensina-aldosterona, la inflamación, el receptor de señales acoplado a la proteína G aberrante y la disfunción endotelial. Común a estos factores encontramos el estrés oxidativo a largo plazo, que causa producción excesiva de especies reactivas del oxígeno (ERO) vascular, disminución de la biodisponibilidad de óxido nítrico y reducción de la capacidad antioxidante2. En el sistema vascular las ERO tienen un papel fisiológico en el control de la función endotelial y el tono vascular, y un rol fisiopatológico relevante en la inflamación, hipertrofia, proliferación, apoptosis, migración, fibrosis y angiogénesis; importantes factores en la remodelación vascular y la disfunción endotelial asociadas a la HTA3.
Casi todos los modelos experimentales de HTA demuestran algunas formas del exceso oxidativo. Como la inhibición de las enzimas generadoras de ERO, los antioxidantes y los depuradores de las ERO reducen la TA mientras que los pro-oxidantes la incrementan, se ha sugerido que las ERO están causalmente asociadas a la HTA, al menos en los modelos animales4. A pesar de que una gran cantidad de datos mantienen el papel del estrés oxidativo en la hipertensión experimental, la evidencia en la HTA humana es poca. La mejor comprensión de estos mecanismos, permitirá contar con conocimientos más profundos que garanticen una mejor prevención, control y la aparición de nuevos objetivos terapéuticos y tratamientos novedosos para esta enfermedad.
Estrés oxidativo (EO): es un estado de la célula en el cual se encuentra alterada la homeostasis oxidación-reducción, es decir, existe un desbalance entre prooxidantes y antioxidantes5.
Radicales libres: son moléculas que en su estructura atómica presentan un electrón desapareado o impar en el orbital externo, que le confiere una configuración especial de gran inestabilidad. Esto lo hace muy inestable, extraordinariamente reactivo y de vida efímera, con una enorme capacidad para combinarse inespecíficamente en la mayoría de los casos, así como con la diversidad de moléculas integrantes de la estructura celular: carbohidratos, lípidos, proteínas y ácidos nucleicos5.
Especies reactivas del oxígeno: son una variedad de radicales libres formados a partir del O2 (Tabla). Las ERO se inactivan por mecanismos enzimáticos o de atrapamiento5.
Tabla. Ejemplos de ERO.

EFECTO NOCIVO DE LAS ERO SOBRE LAS BIOMOLÉCULAS
El daño celular producido por las ERO ocurre sobre diferentes biomoléculas.
Lípidos
Es aquí donde se produce el daño mayor en un proceso que se conoce como peroxidación lipídica, afecta a las estructuras ricas en ácidos grasos poliinsaturados, ya que se altera la permeabilidad de la membrana celular, por lo que se produce edema y muerte celular. La peroxidación lipídica o enranciamiento oxidativo representa una forma de daño tisular que puede ser desencadenado por el oxígeno, el oxígeno atómico (singlete*), el peróxido de hidrógeno y el radical hidroxilo. Los ácidos grasos insaturados son componentes esenciales de las membranas celulares, por lo que se cree son importantes para su funcionamiento normal; sin embargo, son vulnerables al ataque oxidativo iniciado por las ERO6.
Los factores que influyen en la magnitud de la peroxidación lipídica son:
La naturaleza cualitativa y cuantitativa del agente inicializador.
Los contenidos de la membrana en ácidos grasos poliinsaturados y su accesibilidad.
La tensión de oxígeno.
La presencia de hierro.
El contenido celular de antioxidantes (betacarotenos, alfatocoferoles, glutatión).
La activación de enzimas que pueden hacer terminar la cadena de reacción como es el caso de la glutatión peroxidasa.
Una vez que se inicia, el proceso toma forma de "cascada", con producción de ERO que lleva a la formación de peróxidos orgánicos y otros productos, a partir de los ácidos grasos insaturados; una vez formadas, estas ERO son las responsables de los efectos citotóxicos6.
Proteínas
Hay oxidación de un grupo de aminoácidos como fenilalanina, tirosina, histidina y metionina; además se forman entrecruzamientos de cadenas peptídicas, y por último hay formación de grupos carbonilos5.
Ácido desoxirribonucleico (ADN)
Ocurren fenómenos de mutaciones y carcinogénesis, hay pérdida de expresión o síntesis de una proteína por daño a un gen específico, modificaciones oxidativas de las bases, deleciones, fragmentaciones, interacciones estables ADN-proteínas, reordenamientos cromosómicos y desmetilación de citosinas del ADN que activan genes. El daño se puede realizar por la alteración (inactivación/pérdida de algunos genes supresores de tumores que pueden conducir a la iniciación y progresión de la carcinogénesis). Los genes supresores de tumores pueden ser modificados por un simple cambio en una base crítica de la secuencia del ADN5.
GENERACIÓN VASCULAR DE ERO
Las ERO son producidas como intermediarios de las reacciones de reducción-oxidación formando H2O y O2. La secuencia de la reducción univalente del O2 es7,8:
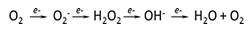
De las ERO generadas en las células vasculares, el O2- (anión superóxido) y el H2O2 (peróxido de hidrógeno) parecen ser particularmente importantes. En los sistemas biológicos, el O2- es de vida media corta y tras su reducción rápida forma H2O2 por la acción de la enzima superóxido dismutasa (SOD), de la cual han sido caracterizadas 3 isoformas en mamíferos: cobre/cinc SOD (SOD1), SOD mitocondrial (SOD2) y SOD extracelular (SOD3)9. La carga de O2- hace difícil que pueda atravesar las membranas celulares, exceptuando la posibilidad que pueda pasar por los canales iónicos. El H2O2 tiene una vida media más larga que el O2-, es relativamente estable y es fácilmente difusible entre las células. La distintas propiedades químicas entre O2- y H2O2 y sus diferentes sitios de distribución sugieren que las distintas especies de ERO activan diversas vías de señales, las cuales guían a la divergencia, oponiendo potencialmente las respuestas biológicas10.
Todos los tipos de células vasculares producen ERO, incluyendo las endoteliales, del músculo liso, los fibroblastos adventiciales, los adipocitos perivasculares y las células fagocíticas (neutrófilos, eosinófilos, monocitos y macrófagos). Estas ERO pueden ser formadas por varias enzimas, incluyendo la xantina óxido-reductasa, la óxido nítrico sintetasa, las enzimas respiratorias mitocondriales y la nicotinamida adenina dinucleótido fosfatasa (NADPH) oxidasa. De estas enzimas las mitocondriales y la NADPH oxidasa parecen ser particularmente importantes en la HTA11-14.
FISIOPATOLOGÍA DE LA HIPERTENSIÓN ASOCIADA AL ESTRÉS OXIDATIVO
Disfunción endotelial
La disfunción endotelial ha sido implicada en la fisiopatología de diferentes formas de enfermedad cardiovascular, incluida la HTA. Esta disfunción puede ser definida como un deterioro de las acciones del endotelio en la vasodilatación, el aumento del estado proinflamatorio y de la actividad protrombótica. Estos fenómenos llevan a un estado de inflamación vascular que puede mediarse parcialmente por las ERO formadas por las células mononucleares activadas15.
El EO vascular y la HTA
El EO constituye un mecanismo común de lesión en muchos tipos de procesos de la enfermedad hipertensiva, y ocurre cuando hay un desequilibrio entre la generación de ERO y los sistemas antioxidantes de defensa del organismo. La familia de ERO comprende muchas moléculas que producen efectos divergentes en la función celular. Precisamente, muchas de estas acciones están asociadas con cambios patológicos observados en la enfermedad cardiovascular. Los efectos de las ERO son mediados través de la regulación redox-sensible de múltiples moléculas de señalización y segundos mensajeros16-18. Varios estudios han demostrado cantidades exageradas de ERO en los pacientes con HTA esencial y varios modelos animales de hipertensión19-21; estos pacientes y los animales de experimentación tienen un estado antioxidante disminuido22, lo que acumula una mayor evidencia de que el EO podría estar involucrado en la hipertensión esencial23. Recientemente fue demostrada una fuerte relación entre la TA y algunos parámetros relacionados con el EO24. Otros estudios experimentales evidenciaron que ratones con deficiencias genéticas en las enzimas generadoras de ERO tienen menor TA en comparación con los ratones de tipo salvaje25,26. Además en cultivos de células de músculo liso vascular provenientes de arterias de ratas y humanos hipertensos, la producción de ERO está aumentada, la señalización redox-dependiente amplificada, y la actividad antioxidante reducida27. Los efectos beneficiosos de agentes antihipertensivos clásicos, como los betabloqueadores, los inhibidores de la enzima conversora de angiotensina (ECA), antagonistas de los receptores de angiotensina II, y bloqueadores de los canales de calcio, son mediados en parte por la disminución del EO vascular28,29.
Las fuentes de ERO en la pared vascular
En los vasos sanguíneos existe una variedad de fuentes enzimáticas y no enzimáticas de ERO. La mejor caracterizada es la NADPH oxidasa. Además, otras varias enzimas pueden contribuir a la generación de ERO, como la xantina oxidasa, la óxido nítrico sintetasa (ONS) y las enzimas mitocondriales.
NADPH Oxidasa
Es la fuente bioquímica primaria de ERO en la vasculatura, particularmente el O2-. También tiene importancia en el riñón, por lo que juega un papel importante en la disfunción renal y el daño vascular en condiciones patológicas30. Este sistema cataliza la reducción de O2 molecular por la NADPH, que funciona como un donador de electrones en la generación de O2-. La NADPH oxidasa es un regulador positivo en la hipertensión por mecanismos humorales y de señales. La angiotensina II es el estímulo más estudiado en la regulación positiva de la NADPH oxidasa, pero la endotelina 1 (ET-1) y la urotensina II también pueden participar en la activación de la NADPH oxidasa, lo que conduce a un incremento de ERO. El efecto más conocido del O2- generado por la NADPH oxidasa es la inactivación del óxido nítrico (ON) en una reacción en que se forma peroxinitrito, que daña la acción vasodilatadora del endotelio y desacopla la ONS endotelial (ONSe), lo que conlleva a una producción adicional de O2- 25,31. En la vasculatura, la activación de la NADPH oxidasa ha sido fuertemente asociada con la HTA32.
Desacoplamiento de la ONSe
La función primaria de la ONSe es la producción de ON que regula la vasodilatación. No obstante, la deficiencia u oxidación de la L-arginina y el tetrahidrobiopterin (BH4), que son 2 cofactores esenciales en el funcionamiento de la ONSe, están asociados con el desacoplamiento de la vía L-arginina- ON, lo que ocasiona una disminución en la formación de ON y el incremento de la generación de O2-, mediada por la ONSe desacoplada.
La NADPH oxidasa es la fuente inicial de ERO. El O2- se combina con el ON formado por la ONSe para formar peroxinitrito33, el que a su vez oxida y desestabiliza la ONSe para producir el O2- adicional34. El O2- también lleva a la oxidación del BH4 que promueve el desacoplamiento de la ONSe y una producción más extensa de ERO.
Xantina oxidasa
La xantina oxidasa es también una fuente importante de ERO en el endotelio vascular35. Cataliza los últimos dos pasos del metabolismo de la purina. Durante este proceso, el O2 se reduce al O2-. Hay evidencia que hace pensar la participación de esta enzima en la HTA. En ratas hipertensas se han demostrado niveles elevados de xantina oxidasa endotelial y aumento de la producción de ERO, que están asociados con el aumento del tono arteriolar30. Además de los efectos en la vasculatura, esta xantina puede tener una función en el daño final de los órganos diana en la HTA36.
Mitocondria
Las mitocondrias son la mayor fuente y blanco de las ERO. Algunos de los O2- producidos en el espacio intermembranal pueden pasar al citoplasma37. La ubiquinona o coenzima Q produce el O2- cuando está parcialmente reducida (en forma de semiquinona), y un antioxidante cuando se encuentra totalmente reducida38. El complejo I produce la mayoría del O2- que se genera en las mitocondrias de los mamíferos. Los complejos II y IV no son sitios normalmente significativos en la producción de ERO. Un desacople moderado del complejo I es muy efectivo en la disminución de la producción de O2- y se ha publicado que existe disminución de la actividad enzimática antioxidante en los pacientes con HTA39.
Papel de los componentes de la pared vascular
El endotelio es sensible a cambios mecánicos y hormonales; en respuesta, libera agentes que regulan la función motora. No hay ninguna duda de que el endotelio tiene un papel protector y regulador a través de la generación de sustancias vasorrelajantes. En circunstancias patológicas el endotelio produce factores vasoconstrictores como ET-1, angiotensina II, urotensina II, aniones superóxidos, prostaglandinas vasoconstrictoras y tromboxano A2, los cuales pueden liberarse y contribuir a los efectos vasoconstrictores paradójicos.
Las células del músculo liso vascular (CMLV) no sólo participan en la regulación a corto plazo del diámetro del vaso sanguíneo y, por consiguiente, de la TA, sino que también intervienen en la adaptación a largo plazo a través de la remodelación estructural. Las ERO median muchos de estos procesos fisiopatológicos (Figura).
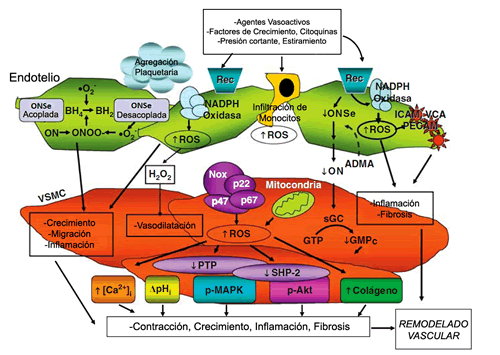
Figura. Remodelado vascular de la hipertensión inducido por el Estrés Oxidativo. La activación de las enzimas generadas de ERO, como la NADPH oxidasa, la actividad simultánea de la ONSe desacoplada y las enzimas mitocondriales en el endotelio y las células musculares lisas vasculares conlleva a la disminución de la producción de ON y la generación aumentada de O2- y H2O2, los cuales influyen sobre moléculas de señalización sensibles a los cambios oxidativos como las MAPKs, PTPs, los canales iónicos, factores de transcripción que inducen la expresión de las moléculas de adhesión proinflamatoria (ICAM), las moléculas de adhesión de la célula vascular y moléculas de adhesión plaquetaria al endotelio (PECAM). Estos procesos conducen al crecimiento vascular, fibrosis, contracción/dilatación, inflamación y la agregación de plaquetas, los cuales sustentan el daño vascular y la remodelación estructural en la hipertensión y otras enfermedades cardiovasculares.
Leyenda: ADMA (Dimetilarginina asimétrica); BH4 (tetrahidrobiopterin); GMPc (guanosina monofosfato cíclico); GTP (guanosina trifosfato); ONSe (óxido nítrico sintetasa endotelial); MAPK (proteínas quinasas activadas por mitógeno); NADPH (nicotinamida adenina dinucleótido fosfatasa); N(G)-dimetil-L-arginina (inhibidor endógeno de la ONS); p (proteína fosforilada); PTP (proteína tirosina fosfatasa); Rec (receptor); sGC (guanililciclasa soluble); SHP-2, (proteína tirosina fosfatasa-2 contiene el dominio SH2); VSMC (células musculares lisas vasculares). Modificada de Touyz RM y Briones AM. Hypertension Research. 2010;5-1410.
La adventicia pueden contribuir a la HTA ya sea al reducir la biodisponibilidad de ON o al participar en la remodelación vascular mediada por las ERO.
Papel de los factores y hormonas vasculares
El ON tiene una importante función como regulador paracrino del tono vascular. Fisiológicamente el ON inhibe la adhesión leucocitaria al endotelio, la proliferación y migración de CMLV, y la agregación plaquetaria, manteniendo de esta forma saludable el endotelio vascular. Por consiguiente tiene muchos efectos beneficiosos. La disminución de la biodisponibilidad de ON en la vasculatura reduce su capacidad vasodilatadora y contribuye a la HTA. La enzima que cataliza la formación de ON a partir de O2 y arginina es la ONS, la cual, de hecho, es una familia entera de enzimas. La ONSe es la isoforma predominante de la ONS en la pared vascular. El estímulo de los receptores por sus agonistas llevan a la rápida activación de la enzima y la tensión cortante y los moduladores alostéricos son importantes reguladores de su actividad40. Además de su acción vasodilatadora y antiproliferativa, el ON tiene una importante función que antagoniza los efectos de la angiotensina II, las endotelinas y las ERO. El ON difunde como un gas al músculo liso subyacente en donde interactúa con diferentes receptores moleculares como la guanililciclasa soluble.
La producción normal de ON tiene una función crucial en el mantenimiento de las condiciones fisiológicas dentro del sistema cardiovascular. La L-arginina, un substrato de la ONSe, parece ser una molécula prometedora en la preservación de la formación de ON; sin embargo, la L-arginina falla en la prevención de cifras altas de TA y el remodelado del ventrículo izquierdo, debido al tratamiento crónico con el éster del metilo de N-nitro-L-arginina que es un inhibidor de la ONSe41. El captopril previene completamente la HTA por deficiencia de ON sin mejorar la actividad de la ONS. El ON también ejerce una retroalimentación negativa sobre la ECA. Los grupos tioles protegen el ON de la oxidación al recoger los deshechos de ERO y formar nitrosotioles, efectos que prolongan la vida media y la duración de la acción del ON42,43.
Los niveles reducidos de ON pueden atribuirse a la elevación de las ERO. El O2- se combina con el ON para formar peroxinitrito que oxida al BH4 y desestabiliza la ONSe para producir más O2- 33,34, lo que aumenta aun más el EO. Por eso, el equilibrio entre el ON y la angiotensina II en el centro vasomotor es muy importante para la regulación del tono simpático.
Sistema de la Renina-Angiotensina
Este sistema tiene un papel importante en el desarrollo de enfermedad cardiovascular. La angiotensina II es un potente péptido vasoactivo que se forma en el lecho vascular rico en ECA. Cuando su producción aumenta sobre los niveles normales, induce la remodelación vascular y la disfunción endotelial, que se asocian a cifras altas de TA, y como es un activador potente de la NADPH oxidasa, contribuye a la producción de ERO44,45. En ratas y ratones donde la HTA es inducida por infusión de angiotensina II, la expresión de las subunidades de la NADPH oxidasa, la actividad oxidante y la generación de ERO están aumentados46. Esta angiotensina II no solamente aumenta la actividad de la NADPH oxidasa, sino que también regula positivamente la actividad de la SOD, posiblemente para compensar los niveles aumentados de ERO. En situaciones donde este efecto compensatorio es eficaz, los niveles de ERO pueden mantenerse normales, incluso en condiciones prooxidantes. No obstante, cuando la producción de ERO se vuelve incontrolable, los mecanismos compensatorios son insuficientes y se desencadenan las consecuencias fisiopatológicas47.
El captopril y el enalapril previnieron los aumentos de la TA en la hipertensión inducida de ratas jóvenes, por inhibición de la ECA. El captopril, probablemente debido al papel antioxidante de su grupo tiol, tuvo un efecto antihipertensivo más eficaz que el enalapril48. En cambio, el ON no sólo antagoniza los efectos de la angiotensina II en el tono vascular, el crecimiento celular y la excreción renal de sodio, sino que también regula negativamente la síntesis de ECA y la expresión de receptores de angiotensina 1. Por tanto, la inhibición de la ECA regula positivamente la expresión de la ONS. La capacidad de la angiotensina II para inducir disfunción endotelial también es debida a su capacidad para regular negativamente la guanililciclasa soluble, lo que ocasiona daño en la señalización de ON/GMPc.
Acetilcolina
En los vasos sanguíneos la acetilcolina induce la dilatación del endotelio por medio de la producción de factores endoteliales, principalmente ON, el cual difunde a la capa de músculo liso vascular subyacente e induce vasorrelajación. La disminución de la biodisponibilidad de ON ocasiona una reducción significativa de la vasodilatación mediada por la acetilcolina48,49 y la consecuencia del aumento global de las ERO es la disminución del ON.
ET-1
Las endotelinas son potentes isopéptidos vasoconstrictores producidos por diferentes tejidos vasculares, entre ellos el endotelio vascular. La ET-1 es la principal endotelina generada en el endotelio y la más importante en el sistema cardiovascular. Cuando es administrada en altas concentraciones, se comporta como un vasoconstrictor potente que es capaz de ejercer una serie de efectos, como alterar la TA. La ET-1 actúa a través de dos receptores: ETA y ETB. El receptor de ETA media la vasoconstricción por vía de la activación de la NADPH oxidasa, la xantina oxidasa, la lipoxigenasa, el desacoplamiento de la ONSe y las enzimas de las cadenas respiratorias mitocondriales. El receptor de ETB induce la relajación de las células endoteliales50. Muchos factores que normalmente estimulan la síntesis de ET-1, (por ejemplo: trombina y angiotensina II) también causan la elevación de vasodilatadores, como la prostaciclina (PGI2) y el ON, o ambos, que se oponen a la función vasoconstrictora de la ET-1. Por eso se ha informado que la HTA esencial se caracteriza por un aumento del tono vasoconstrictor mediado por la ET-1, además de la disminución de ETB como consecuencia de la producción disminuida de ON, o su pobre biodisponibilidad.
Urotensina-II
La urotensina II es un poderoso péptido vasoactivo51 y es, de hecho, el vasoconstrictor más potente que se ha identificado. Funciona a través de la activación de la NADPH oxidasa. El papel de la urotensina II en la HTA aún no es bien comprendido, pues la respuesta vasoconstrictora parece ser variable y muy dependiente del lecho vascular; no obstante, la vasoconstricción no es su único efecto ya que se han identificado receptores de urotensina II en otros órganos52,53. Este péptido también parece funcionar como un vasodilatador potente en algunos vasos aislados54.
Norepinefrina
Las CMLV son inervadas principalmente por el sistema nervioso simpático a través de los receptores adrenérgicos. Tres tipos de receptores están presentes en las CMLV: α1, α2 y β2. La norepinefrina estimula la proliferación del músculo liso vascular. En adición, la sobreexpresión del ON aumenta las cifras de PA debido a la activación del sistema nervioso simpático mediado por el incremento del EO16.
Prostaglandinas
La PGI2 es otro vasodilatador producido en el endotelio que relaja el músculo liso vascular. Es liberada en cantidades superiores en respuesta a diferentes compuestos como la trombina, el ácido araquidónico, la histamina y la serotonina. La enzima prostaglandina H2 sintetasa usa el ácido araquidónico como substrato para producir la prostaglandina H2, la cual se convierte en moléculas vasoactivas como la PGI2. La isoforma de la enzima prostaglandina H2 sintetasa 2, puede participar en el trastorno vascular bajo condiciones de EO; así, el peroxinitrito inhibe la actividad enzimática de la PGI2 sintetasa y afecta la vasodilatación mediada por la PGI2.
Homocisteína
Esta molécula puede tener un papel importante en la patogénesis de la HTA esencial. La homocisteinemia elevada disminuye la vasodilatación del ON, aumenta el EO, estimula la proliferación del músculo liso vascular y altera las propiedades elásticas de la pared del vaso. Por tanto la homocisteína contribuye a la elevación de la TA. Adicionalmente, sus niveles elevados podrían ocasionar daño oxidativo del endotelio55. La corrección de la homocisteinemia producida por la administración de las vitaminas B6, B12 y ácido fólico, podría ser una terapia útil en la HTA6; sin embargo, son necesarios más ensayos controlados y aleatorizados para establecer la eficacia de estos agentes terapéuticos.
RIÑÓN Y SISTEMA NERVIOSO CENTRAL
Hasta aquí se ha discutido la importancia de las ERO en los vasos sanguíneos y su relación con la HTA; sin embargo, es importante también enfatizar la evidencia de que estímulos hipertensivos, como la ingestión elevada de sal y la angiotensina II, no sólo promueven la producción de ERO a este nivel, sino también en el riñón y en el sistema nervioso central. Además, cada uno de estos sitios también contribuye a la HTA, o las secuelas adversas de esta enfermedad56.
Importancia del EO en el riñón
La evidencia hace pensar que las ERO tienen un papel clave en los procesos fisiopatológicos de varias enfermedades renales, las que son consideradas causas y consecuencias de HTA. Con respecto a las alteraciones glomerulares, las ERO producen lipoproteínas gromerulopáticas y otras lesiones inflamatorias glomerulares57. Un estudio reciente demostró que algunos conglomerados de lípidos producen la activación de la NADPH oxidasa y la producción de ERO, lo que es un mecanismo molecular importante que estimula la homocisteína, la cual favorece el daño oxidativo de los podocitos. Este daño puede representar un fenómeno temprano que inicia la glomeruloesclerosis durante la hiperhomocisteinemia58. Uno de los mecanismos subyacentes de la lesión túbulo-intersticial, mediada por las ERO, es la exposición de células tubulares a las LDL, que pueden producir alteraciones en los túbulos del intersticio debido a la producción de ERO por la NADPH oxidasa59. La angiotensina II no sólo tienen un papel patogénico en la progresión de lesión túbulo-intersticial, sino también en la nefropatía obstructiva60,61; además, activa la NADPH oxidasa y genera O2- que ocasiona la hipertrofia de las células tubulares renales62.
Hay hallazgos que sugieren que una dieta alta en grasas induce la inflamación renal y el aumento de la TA por medio de las ERO en la HTA de las ratas63. Adicionalmente, el síndrome metabólico es un factor de riesgo para la insuficiencia renal crónica (IRC), independiente de la diabetes y la HTA, probablemente debido a la influencia de las ERO. El inicio y el mantenimiento del daño renal pueden empeorar el síndrome metabólico, así como la HTA64.
Hay varios mecanismos de EO involucrados en la disfunción endotelial de la IRC65, donde las ERO están elevadas y se relacionan con la reactividad vascular del endotelio y la TA sistólica66. Los niveles altos de ERO y de dimetilarginina asimétrica han sido reconocidos como nuevos factores de riesgo de la disfunción endotelial67. Además, se han sido encontrado altos niveles de esta dimetilarginina en la IRC que están asociados con el aumento del espesor de la íntima y la media vascular, y el incremento de accidentes cardiovasculares68.
Importancia del EO en el sistema nervioso central
Además del riñón y los vasos sanguíneos, el sistema nervioso simpático, que a su vez es regulado por el sistema nervioso central, está involucrado en la patogénesis de la HTA69. Recientes estudios sugieren que el aumento del estímulo simpático central incrementa la TA70. Hay también pruebas de que la generación aumentada de ERO en el cerebro contribuye a los mecanismos neurales involucrados en la HTA de las ratas71.
La médula ventrolateral rostral es el mayor centro vasomotor, y es esencial en el mantenimiento del tono vascular basal72,73. Algunos resultados indican que el incremento de las ERO a este nivel aumenta el estímulo vasomotor de la HTA espontánea en ratas y por eso contribuye a los mecanismos neurales de la HTA a través de la activación del sistema nervioso simpático72. El núcleo paraventricular del hipotálamo es, tal vez, el más fuertemente asociado a los mecanismos neurales de la hipertensión relacionada con las ERO74. Hay evidencia de que otras regiones del cerebro están igualmente involucradas en este tipo de HTA. Estas investigaciones sugieren que la producción aumentada de O2- intracelular en el órgano subfornical es crítica en el desarrollo de la HTA inducida por la angiotensina II75.
Conclusiones
Numerosos datos confirman la importancia de las ERO en el control de la función vascular, por medio de la regulación de la función endotelial y el tono vascular a través del estricto control de las vías de señalización redox-sensibles. Las ERO son mediadores de la mayoría de los vasoconstrictores fisiológicos que aumentan la concentración de calcio intracelular. El O2- reduce la biodisponibilidad de ON y desacopla la ONSe, lo que aumenta aún más las concentraciones de O2-. La producción/degradación descontrolada de las ERO provoca el EO, que induce daño cardiovascular, renal y neural, y se asocia al aumento de la TA. Aunque el daño oxidativo no es la única causa de HTA, facilita y aumenta la elevación de la TA en presencia de otros factores prohipertensivos. Los resultados obtenidos de los estudios experimentales y en animales sugieren el papel del EO en la patogénesis de la HTA, posiblemente a través de la activación de enzimas oxidantes, donde las NADPH oxidasas y las mitocondriales tienen un importante desempeño. Desde el punto de vista clínico, se necesita que estos datos sobre el papel causal de las ERO en la HTA humana continúen en investigación.
Nota del editor
* Oxígeno singlete, este término viene del inglés singlet oxygen. Una traducción acertada podría ser oxígeno atómico, pues es un átomo de oxígeno en un estado excitado. Es el nombre común utilizado para las formas energéticamente excitadas del oxígeno molecular (O2), con dos electrones apareados en los orbitales de energía más alta (orbital antienlazante), y es menos estable que el oxígeno normal. El oxígeno atómico no es un radical, sino una especie activada.
Referencias bibliográficas
Kakar P, Lip GY. Towards understanding the aetiology and pathophysiology of human hypertension: where are we now? J Hum Hypertens. 2006;20(11):833-6.
Viel EC, Lemarié CA, Benkirane K, Paradis P, Schiffrin EL. Immune regulation and vascular inflammation in genetic hypertension. Am J Physiol Heart Circ Physiol. 2010;298(3):938-44.
Vaziri ND, Rodríguez-Iturbe B. Mechanisms of disease: oxidative stress and inflammation in the pathogenesis of hypertension. Nat Clin Pract Nephrol. 2006;2(10):582-93.
Kagota S, Tada Y, Kubota Y, Nejime N, Yamaguchi Y, Nakamura K, et al. Peroxynitrite is involved in the dysfunction of vasorelaxation in SHR/NDmcr-cp rats, spontaneously hypertensive obese rats. J Cardiovasc Pharmacol. 2007;50(6):677-85.
Venereo JR. Daño oxidativo, radicales libres y antioxidantes. Rev Cubana Med Milit. 2002;31(2):126-33.
Jerlich A, Pitt AR, Schaur RJ, Spickett CM. Pathway of phospholipid oxidation by HOCl in human LDL detected by LC-MS. Free Radic Biol Med. 2000;28(5):673-82.
Lavi S, Yang EH, Prasad A, Mathew V, Barsness GW, Rihal CS, et al. The interaction between coronary endothelial dysfunction, local oxidative stress, and endogenous nitric oxide in humans. Hypertension. 2008;51(1):127-33.
Johnson F, Giulivi C. Superoxide dismutases and their impact upon human health. Mol Aspects Med. 2005;26(4-5):340-52.
Mendez JI, Nicholson WJ, Taylor WR. SOD isoforms and signaling in blood vessels: evidence for the importance of ROS compartmentalization. Arterioscler Thromb Vasc Biol. 2005;25(5):887-8.
Touyz RM, Briones AM. Reactive oxygen species and vascular biology: implications in human hypertension. Hypertension Res. 2011;34(1):5-14.
Nishino T, Okamoto K, Eger BT, Pai EF, Nishino T. Mammalian xanthine oxidoreductase - mechanism of transition from xanthine dehydrogenase to xanthine oxidase. FEBS J. 2008;275(13):3278-89.
Moens AL, Kass DA. Tetrahydrobiopterin and cardiovascular disease. Arterioscler Thromb Vasc Biol. 2006;26(11):2439-44.
Liu Y, Zhao H, Li H, Kalyanaraman B, Nicolosi AC, Gutterman DD. Mitochondrial sources of H2O2 generation play a key role in flow-mediated dilation in human coronary resistance arteries. Circ Res. 2003;93(9):573-80.
DeLano FA, Parks DA, Ruedi JM, Babior BM, Schmid-Schönbein GW. Microvascular display of xanthine oxidase and NADPH oxidase in the spontaneously hypertensive rat. Microcirculation 2006;13(7):551-66.
Badimón L, Martínez-González J. Disfunción endotelial. Rev Esp Cardiol. 2006;6(Supl A):21-30.
Kimura S, Zhang GX, Nishiyama A, Shokoji T, Yao L, Fan YY, et al. Mitochondria-derived reactive oxygen species and vascular MAP kinases: comparison of angiotensin II and diazoxide. Hypertension. 2005;45(3):438-44.
Hool LC, Corry B. Redox control of calcium channels: from mechanisms to therapeutic opportunities. Antioxid Redox Signal. 2007;9(4):409-35.
Yoshioka J, Schreiter ER, Lee RT. Role of thioredoxin in cell growth through interactions with signaling molecules. Antioxid Redox Signal. 2006;8(11):2143-51.
Redón J, Oliva MR, Tormos C, Giner V, Chaves J, Iradi A, et al. Antioxidant activities and oxidative stress byproducts in human hypertension. Hypertension. 2003;41(5):1096-101.
Tanito M, Nakamura H, Kwon YW, Teratani A, Masutani H, Shioji K, et al. Enhanced oxidative stress and impaired thioredoxin expression in spontaneously hypertensive rats. Antioxid Redox Signal. 2004;6(1):89-97.
Touyz RM. Reactive oxygen species, vascular oxidative stress, and redox signaling in hypertension: what is the clinical significance? Hypertension. 2004;44(3):248-52.
Briones AM, Touyz RM. Oxidative stress and hypertension: current concepts. Curr Hypertens Rep. 2010;12(2):135-42.
Bengtsson SH, Gulluyan LM, Dusting GJ, Drummond GR. Novel isoforms of NADPH oxidase in vascular physiology and pathophysiology. Clin Exp Pharmacol Physiol. 2003;30(11):849-54.
Rodrigo R, Prat H, Passalacqua W, Araya J, Guichard C, Bächler JP. Relationship between oxidative stress and essential hypertension. Hypertens Res. 2007;30(12):1159-67.
Landmesser U, Dikalov S, Price SR, McCann L, Fukai T, Holland SM, et al. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Invest. 2003;111(8):1201-9.
Gavazzi G, Banfi B, Deffert C, Fiette L, Schappi M, Herrmann F, et al. Decreased blood pressure in NOX1-deficient mice. FEBS Lett. 2006;580(2):497-504.
Touyz RM, Schiffrin EL. Increased generation of superoxide by angiotensin II in smooth muscle cells from resistance arteries of hypertensive patients: role of phospholipase D-dependent NAD(P)H oxidase-sensitive pathways. J Hypertens. 2001;19(7):1245-54.
Ghiadoni L, Magagna A, Versari D, Kardasz I, Huang Y, Taddei S, et al. Different effect of antihypertensive drugs on conduit artery endothelial function. Hypertension. 2003;41(6):1281-6.
Yoshida J, Yamamoto K, Mano T, Sakata Y, Nishikawa N, Nishio M, et al. AT1 receptor blocker added to ACE inhibitor provides benefits at advanced stage of hypertensive diastolic heart failure. Hypertension. 2004;43(3):686-91.
Feairheller DL, Brown MD, Park JY, Brinkley TE, Basu S, Hagberg JM, et al. Exercise training, NADPH oxidase p22phox gene polymorphisms, and hypertension. Med Sci Sports Exerc. 2009;41(7):1421-8.
Zou MH, Cohen R, Ullrich V. Peroxynitrite and vascular endothelial dysfunction in diabetes mellitus. Endothelium. 2004;11(2):89-97.
Lassègue B, Clempus RE. Vascular NAD(P)H oxidases: specific features, expression, and regulation. Am J Physiol Regul Integr Comp Physiol. 2003;285(2):277-97.
Kuzkaya N, Weissmann N, Harrison DG, Dikalov S. Interactions of peroxynitrite, tetrahydrobiopterin, ascorbic acid, and thiols: implications for uncoupling endothelial nitric-oxide synthase. J Biol Chem. 2003;278(25):22546-54.
Laursen JB, Somers M, Kurz S, McCann L, Warnholtz A, Freeman BA, et al. Endothelial regulation of vasomotion in apoE-deficient mice: implications for interactions between peroxynitrite and tetrahydrobiopterin. Circulation. 2001;103(9):1282-8.
Viel EC, Benkirane K, Javeshghani D, Touyz RM, Schiffrin EL. Xanthine oxidase and mitochondria contribute to vascular superoxide anion generation in DOCA-salt hypertensive rats. Am J Physiol Heart Circ Physiol. 2008;295(1):281-8.
Laakso JT, Teräväinen TL, Martelin E, Vaskonen T, Lapatto R. Renal xanthine oxidoreductase activity during development of hypertension in spontaneously hypertensive rats. J Hypertens. 2004;22(7):1333-40.
Han D, Antunes F, Canali R, Rettori D, Cadenas E. Voltage-dependent anion channels control the release of the superoxide anion from mitochondria to cytosol. J Biol Chem. 2003;278(8):5557-63.
Eto Y, Kang D, Hasegawa E, Takeshige K, Minakami S. Succinate-dependent lipid peroxidation and its prevention by reduced ubiquinone in beef heart submitochondrial particles. Arch Biochem Biophys. 1992;295(1):101-6.
Zhou L, Xiang W, Potts J, Floyd M, Sharan C, Yang H, et al. Reduction in extracellular superoxide dismutase activity in African-American patients with hypertension. Free Radic Biol Med. 2006;41(9):1384-91.
Michel JB, Feron O, Sase K, Prabhakar P, Michel T. Caveolin versus calmodulin. Counterbalancing allosteric modulators of endothelial nitric oxide synthase. J Biol Chem. 1997;272(41):25907-12.
Simko F, Luptak I, Matuskova J, Krajcirovicova K, Sumbalova Z, Kucharska J, et al. L-arginine fails to protect against myocardial remodelling in L-NAME-induced hypertension. Eur J Clin Invest. 2005;35(6):362-8.
Zhang Y, Hogg N. S-Nitrosothiols: cellular formation and transport. Free Radic Biol Med. 2005;38(7):831-8.
Sládková M, Kojsová S, Jendeková L, Pechánová O. Chronic and acute effects of different antihypertensive drugs on femoral artery relaxation of L-NAME hypertensive rats. Physiol Res. 2007;56(Suppl 2):85-91.
Touyz RM. Reactive oxygen species and angiotensin II signaling in vascular cells - implications in cardiovascular disease. Braz J Med Biol Res. 2004;37(8):1263-73.
Hitomi H, Kiyomoto H, Nishiyama A. Angiotensin II and oxidative stress. Curr Opin Cardiol. 2007;22(4):311-5.
Landmesser U, Cai H, Dikalov S, McCann L, Hwang J, Jo H, et al. Role of p47(phox) in vascular oxidative stress and hypertension caused by angiotensin II. Hypertension. 2002;40(4):511-5.
Taniyama Y, Griendling KK. Reactive oxygen species in the vasculature: molecular and cellular mechanisms. Hypertension. 2003;42(6):1075-81.
Pechánová O. Contribution of captopril thiol group to the prevention of spontaneous hypertension. Physiol Res. 2007;56(Suppl 2):41-8.
Bitar MS, Wahid S, Mustafa S, Al-Saleh E, Dhaunsi GS, Al-Mulla F. Nitric oxide dynamics and endothelial dysfunction in type II model of genetic diabetes. Eur J Pharmacol. 2005;511(1):53-64.
Gomez-Alamillo C, Juncos LA, Cases A, Haas JA, Romero JC. Interactions between vasoconstrictors and vasodilators in regulating hemodynamics of distinct vascular beds. Hypertension. 2003;42(4):831-6.
Djordjevic T, BelAiba RS, Bonello S, Pfeilschifter J, Hess J, Görlach A. Human urotensin II is a novel activator of NADPH oxidase in human pulmonary artery smooth muscle cells. Arterioscler Thromb Vasc Biol. 2005;25(3):519-25.
Matsushita M, Shichiri M, Imai T, Iwashina M, Tanaka H, Takasu N, et al. Co-expression of urotensin II and its receptor (GPR14) in human cardiovascular and renal tissues. J Hypertens. 2001;19(12):2185-90.
Jégou S, Cartier D, Dubessy C, Gonzalez BJ, Chatenet D, Tostivint H, et al. Localization of the urotensin II receptor in the rat central nervous system. J Comp Neurol. 2006;495(1):21-36.
Stirrat A, Gallagher M, Douglas SA, Ohlstein EH, Berry C, Kirk A, et al. Potent vasodilator responses to human urotensin-II in human pulmonary and abdominal resistance arteries. Am J Physiol Heart Circ Physiol. 2001;280(2):925-8.
Rodrigo R, Passalacqua W, Araya J, Orellana M, Rivera G. Homocysteine and essential hypertension. J Clin Pharmacol. 2003;43(12):1299-306.
Harrison DG, Gongora MC. Oxidative stress and hypertension. Med Clin North Am. 2009;93(3):621-35.
Rodrigo R, Rivera G. Renal damage mediated by oxidative stress: a hypothesis of protective effects of red wine. Free Radic Biol Med. 2002;33(3):409-22.
Zhang C, Hu JJ, Xia M, Boini KM, Brimson C, Li PL. Redox signaling via lipid raft clustering in homocysteine-induced injury of podocytes. Biochim Biophys Acta. 2010;1803(4):482-91.
Piccoli C, Quarato G, D´Aprile A, Montemurno E, Scrima R, Ripoli M, et al. Native LDL-induced oxidative stress in human proximal tubular cells: multiple players involved. J Cell Mol Med. 2009;15(2):375-95.
Klahr S. Urinary tract obstruction. Semin Nephrol. 2001;21(2):133-45.
Grande MT, Pérez-Barriocanal F, López-Novoa JM. Role of inflammation in tubulo-interstitial damage associated to obstructive nephropathy. J Inflamm (Lond) [Internet]. 2010 [Citado 2013 Abr 13];22(7):19. Disponible en: http://www.journal-inflammation.com/content/7/1/19
Sachse A, Wolf G. Angiotensin II-induced reactive oxygen species and the kidney. J Am Soc Nephrol. 2007;18(9):2439-46.
Chung S, Park CW, Shin SJ, Lim JH, Chung HW, Youn DY, et al. Tempol or candesartan prevents high-fat diet-induced hypertension and renal damage in spontaneously hypertensive rats. Nephrol Dial Transplant. 2010;25(2):389-99.
Guarnieri G, Zanetti M, Vinci P, Cattin MR, Pirulli A, Barazzoni R. Metabolic syndrome and chronic kidney disease. J Ren Nutr. 2010;20(Suppl 5):19-23.
Malyszko J. Mechanism of endothelial dysfunction in chronic kidney disease. Clin Chim Acta. 2010;411(19-20):1412-20.
Costa-Hong V, Bortolotto LA, Jorgetti V, Consolim-Colombo F, Krieger EM, Lima JJ. Oxidative stress and endothelial dysfunction in chronic kidney disease. Arq Bras Cardiol. 2009;92(5):381-6.
Zoccali C, Bode-Böger S, Mallamaci F, Benedetto F, Tripepi G, Malatino L, et al. Plasma concentration of asymmetrical dimethylarginine and mortality in patients with end-stage renal disease: a prospective study. Lancet 2001;358(9299):2113-7.
Nanayakkara PW, Teerlink T, Stehouwer CD, Allajar D, Spijkerman A, Schalkwijk C, et al. Plasma asymmetric dimethylarginine (ADMA) concentration is independently associated with carotid intima-media thickness and plasma soluble vascular cell adhesión molecule-1 (sVCAM-1) concentration in patients with mild-to-moderate renal failure. Kidney Int. 2005;68(5):2230-6.
Grassi G. Assessment of sympathetic cardiovascular drive in human hypertension: achievements and perspectives. Hypertension. 2009;54(4):690-7.
Guyenet PG. The sympathetic control of blood pressure. Nat Rev Neurosci. 2006;7(5):335-46.
Kishi T, Hirooka Y, Kimura Y, Ito K, Shimokawa H, Takeshita A. Increased reactive oxygen species in rostral ventrolateral medulla contribute to neural mechanisms of hypertension in stroke-prone spontaneously hypertensive rats. Circulation. 2004;109(19):2357-62.
Hirooka Y, Sagara Y, Kishi T, Sunagawa K. Oxidative stress and central cardiovascular regulation. Pathogenesis of hypertension and therapeutic aspects. Circ J. 2010;74(5):827-35.
Sved AF, Ito S, Sved JC. Brainstem mechanisms of hypertension: role of the rostral ventrolateral medulla. Curr Hypertens Rep. 2003;5(3):262-8.
Oliveira-Sales EB, Nishi EE, Carillo BA, Boim MA, Dolnikoff MS, Bergamaschi CT, et al. Oxidative stress in the sympathetic premotor neurons contributes to sympathetic activation in renovascular hypertension. Am J Hypertens. 2009;22(5):484-92.
Zimmerman MC, Lazartigues E, Sharma RV, Davisson RL. Hypertension caused by angiotensin II infusion involves increased superoxide production in the central nervous system. Circ Res. 2004;95(2):210-6.
Recibido: 28 de diciembre de 2012
Modificado: 4 de mayo de 2013
Aceptado: 13 de junio de 2013
Subir

















