



CorSalud 2012 Oct-Dic;4(4):300-306
CASO CLÍNICO
ATIPICIDADES EN UN CASO CON DISPLASIA ARRITMOGÉNICA DEL VENTRÍCULO DERECHO O ENFERMEDAD DE UHL
MSc.Dr. Luis M. Morales Pérez1, Dr. Omar R. González Greck2, MSc.Dra. Ana M. Jerez Castro3, MSc.Dr. Eliezer San Román García4 y Dr. Aníbal González Trujillo5
______________
Especialista de I Grado en Medicina General Integral y en Cardiología. Máster en Urgencias Médicas y en Enfermedades Infecciosas. Diplomado en Cuidados Intensivos y Emergencias.
Especialista de I Grado en Medicina Interna y de II Grado en Cardiología. Profesor e Investigador Auxiliar. Jefe de Terapia Intensiva Cardioquirúrgica.
Especialista de I Grado en Medicina Interna y en Cardiología. Máster en Urgencias Médicas y en Enfermedades Infecciosas. Diplomado en Cuidados Intensivos y Emergencias.
Especialista de I Grado en Medicina General Integral y en Cardiología. Máster en Urgencias Médicas. Diplomado en Cuidados Intensivos y Emergencias.
Especialista de I Grado en Medicina General Integral y en Cardiología.
Departamento de Terapia Intensiva Cardioquirúrgica. Instituto Nacional de Cardiología y Cirugía Cardiovascular, La Habana, Cuba
Correspondencia: LM Morales Pérez. Instituto de Cardiología y Cirugía Cardiovascular. Terapia Intensiva Cardioquirúrgica. Calle 17 N° 702. El Vedado, CP 10400. La Habana, Cuba. Correo Electrónico: luism.morales@infomed.sld.cu
Resumen
La displasia arritmogénica del ventrículo derecho es una miocardiopatía caracterizada por arritmias ventriculares malignas y anomalías estructurales progresivas, que afectan primariamente al ventrículo derecho. Se presenta por una sustitución progresiva parcial o masiva del miocardio por tejido adiposo o fibroadiposo. La enfermedad de Uhl puede ser una manifestación extrema y generalizada de la displasia arritmogénica del ventrículo derecho, trastorno congénito muy poco frecuente con ausencia de miocardio ventricular derecho, por lo que sus paredes son delgadas como el papel. Se comenta el caso de un paciente masculino de 56 años que presentó pérdida de conocimiento y se le realizó el diagnóstico clínico y ecocardiográfico. Se discuten las características clínicas, el diagnóstico y la conducta a seguir ante esta cardiopatía potencialmente letal en pacientes que sufren síncope, taquicardia ventricular o parada cardíaca.
Palabras clave: Displasia ventricular derecha arritmogénica, Cardiomiopatía
Abstract
Arrhythmogenic right ventricular dysplasia is a cardiomyopathy characterized by malignant ventricular arrhythmias and progressive structural abnormalities, affecting primarily the right ventricle. It appears due to a partial or massive progressive replacement of the myocardium by fibroadipose or adipose tissue. Uhl's disease may be an extreme and widespread manifestation of arrhythmogenic right ventricular dysplasia, a rare congenital disorder with absence of right ventricular myocardium, so that its walls are paper thin. The case of a 56 year old male patient who had loss of consciousness and underwent clinical and echocardiographic diagnosis is presented. The clinical features, diagnosis and action to take against this potentially fatal heart disease in patients with syncope, ventricular tachycardia or cardiac arrest are discussed.
Key words: Arrhythmogenic right ventricular dysplasia, Cardiomyopathy
Introducción
La displasia arritmogénica del ventrículo derecho (DAVD) es una miocardiopatía caracterizada por anomalías estructurales progresivas que afectan primariamente al ventrículo derecho (VD) y producen arritmias ventriculares1,2. Este raro trastorno fue descrito en 1977 por Fontaine y colaboradores3. La prevalencia general es difícil de estimar, pues muchos casos son diagnosticados posmortem; sin embargo, es la causa del 3 al 4 % de las muertes en deportistas, y de un 5 % de todas las muertes súbitas antes de los 65 años de edad4. La degeneración miocárdica puede extenderse al tabique interventricular y al ventrículo izquierdo, sobre todo en las fases avanzadas de la enfermedad5. La DAVD puede presentarse en formas esporádicas y familiares. La predisposición familiar fue descrita en 1982 por Marcus y colaboradores6-7, alrededor del 30 % de los pacientes diagnosticados refieren historia familiar4,8 y se han identificado varias alteraciones genéticas responsables4,5,9. La enfermedad se caracteriza por una sustitución progresiva parcial o masiva del miocardio por tejido adiposo o fibroadiposo. Esta infiltración constituye un sustrato para la inestabilidad eléctrica y lleva a diversas arritmias, que van desde las extrasístoles ventriculares aisladas hasta las taquicardias ventriculares (TV) mantenidas o la fibrilación ventricular (FV)2,5,9. La enfermedad de Uhl puede ser una manifestación extrema y generalizada de la DAVD, trastorno congénito muy poco frecuente con ausencia de miocardio ventricular derecho, por lo que sus paredes son delgadas como el papel7-8,10. Gaffney y colaboradores11, postulan que la anomalía de Uhl y la DAVD serían manifestaciones de un único, y presumiblemente congénito proceso patológico: el síndrome del VD apergaminado11.
La ecocardiografía es una técnica incruenta y representa el método de primera línea para evaluar a los pacientes con sospecha diagnóstica de displasia y para el pesquisaje familiar12. Los criterios diagnósticos de la DAVD se dividen en mayores y menores13.
A continuación se describe el caso de un paciente que fue diagnosticado en la Unidad de Cuidados Intensivos Cardioquirúrgicos que había ingresado por un episodio sincopal. Este caso fue publicado en la Revista de la Federación Argentina de Cardiología10 en la sección Imágenes en Cardiología, pero por su importancia y relevancia se ha decidido presentarlo también como un caso clínico.
Caso clínico
Se presenta el atípico caso de un hombre de 56 años de edad con antecedentes patológicos familiares de muerte súbita prematura (<35 años), y personales de palpitaciones, fatiga, dolor torácico atípico y varios episodios de síncope desencadenados por el esfuerzo (las primeras manifestaciones comenzaron entre los 15 y los 35 años). El motivo de su ingreso en la Unidad de Cuidados Intensivos Cardioquirúrgicos fue un episodio sincopal precedido de palpitaciones; negó dolor torácico o disnea. En el examen físico se encontró un paciente consciente, eupneico, hipotenso y taquicárdico (TA 75/55 mm/Hg y frecuencia cardíaca de 160 por minuto), con saturación periférica de oxígeno de 97 % (a aire ambiental), y pulsos periféricos filiformes con discreta ingurgitación yugular. La auscultación cardíaca reveló un corazón arrítmico, muy taquicárdico, e impresionaba la presencia de cuarto ruido derecho con tonos hipofonéticos. La auscultación pulmonar era normal y el abdomen timpánico difuso, doloroso a la palpación en cuadrantes superiores, y sin signos de irritación peritoneal; se evidenció una hepatomegalia dolorosa de aproximadamente 3 cm, con predominio del lóbulo derecho. Los miembros inferiores presentaban un edema leve. Los análisis hematológicos y bioquímicos no mostraron alteraciones significativas. En la radiografía de tórax se observó un corazón dilatado con predominio de cavidades derechas. En el electrocardiograma (ECG) se constató una TV mantenida con morfología de bloqueo de rama izquierda del Haz de His (BRIHH), y un eje del QRS indeterminado, sin signos de isquemia miocárdica aguda. En el ecocardiograma transtorácico se demostró la presencia de un VD dilatado con disfunción sistólica grave y fracción de eyección ventricular derecha (FEVD) < 30 %, determinada por el método de Simpsom (Figura 1). Se constataron además zonas disquinéticas y aneurismas regionales en el VD con aumento del diámetro telediastólico (Figura 2). La morfología ventricular recuerda el papel de cebolla y se le conoce como VD apergaminado. Se revirtió la arritmia ventricular con una cardioversión farmacológica (amiodarona), para mejorar su estado hemodinámico. Se administró terapia con volumen, sin lograr una respuesta adecuada de la tensión arterial, por lo que se inició tratamiento vasopresor e inotrópico. El paciente continuó hipotenso y falleció 12 días después de su ingreso por choque circulatorio.
El estudio patológico no se pudo realizar, pues los familiares no dieron el consentimiento. Se concluye el caso como una DAVD o enfermedad de Uhl, según los criterios de Marcus y colaboradores13.
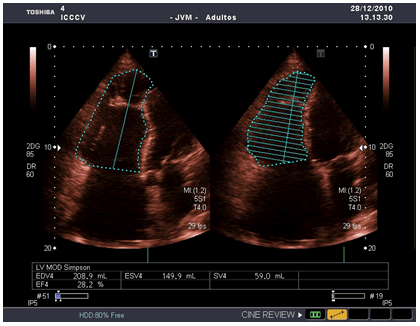
Figura 1. Ecocardiograma transtorácico. Vista apical de 4 cámaras. Determinación de la función ventricular derecha FEVD < 30 % (Método de Simpsom). Imagen tomada de (fuente original) Rev Fed Arg Cardiol. 2012; 41(1): 59-60.
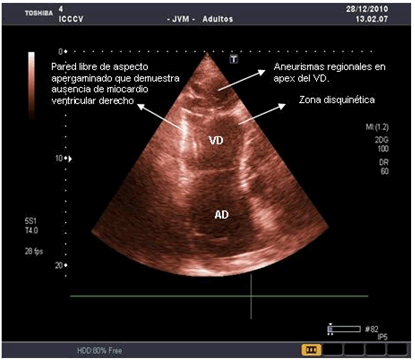
Figura 2. Ecocardiograma transtorácico. Vista apical. Ausencia de miocárdio del VD, se observan paredes de aspecto apergaminado, donde se evidencian zonas disquinéticas y aneurismas regionales en el apex. VD: ventrículo derecho, AD: aurícula derecha. Imagen tomada de (fuente original) Rev Fed Arg Cardiol. 2012; 41(1): 59-60.
Comentario
La DAVD es una miocardiopatía cuya anomalía estructural fundamental es la degeneración miocárdica del ventrículo derecho, que en estadios avanzados de la enfermedad puede extenderse al ventrículo izquierdo. La prevalencia de la enfermedad en la población general se ha estimado en valores que van de 1/2.000 a 1/10.000. El 80 % de los casos se diagnostica en pacientes de edad inferior a los 40 años. Debe sospecharse en todos los pacientes jóvenes con un corazón aparentemente normal que sufren un síncope, TV o parada cardíaca5.
La DAVD es un trastorno heredable. Hay una clara incidencia familiar (30-50 % de los casos), con un patrón de transmisión autosómico dominante, diversos grados de penetración y una expresión fenotípica polimórfica. Se ha descrito también una forma autosómica recesiva7.
La primera mutación causante de una DAVD no sindrómica fue descrita por Rampazzo et al.14 en el año 2002, dicha mutación se identificó en el gen de la desmoplaquina, que codifica un componente del desmosoma. En el 2004, Gerull et al.15 describieron 25 mutaciones del gen desmosómico cardíaco de la placofilina 2. Actualmente se considera que la disfunción desmosómica es la vía final común en la patogenia de la DAVD. Se han localizado en el mapa cromosómico diferentes variantes genéticas de DAVD y se han descrito más de 140 mutaciones causantes de la enfermedad, la mayoría de ellas correspondientes a genes que codifican proteínas desmosómicas15. La integridad de los desmosomas es necesaria para mantener la función normal de las uniones estrechas como canales intercelulares encargados del acoplamiento eléctrico y los mecanismos de señalización en la regulación del crecimiento, la diferenciación y el desarrollo celulares. Las mutaciones genéticas, causa de la DAVD, dan lugar a una haploinsuficiencia y una reducción de la expresión de las proteínas desmosómicas, que pueden predisponer a la rotura de los contactos celulares mecánicos, posiblemente desencadenados por una tensión mecánica del VD (como ocurre durante el ejercicio o la actividad deportiva). La degeneración y la muerte de los miocardiocitos son la consecuencia anátomo-patológica de estas mutaciones de proteínas de adhesión, con la consiguiente sustitución progresiva por tejido adiposo y fibroadiposo16.
La DAVD debe diferenciarse de la enfermedad de Uhl, un trastorno congénito muy poco frecuente con ausencia de miocardio ventricular derecho, con lo que la pared del VD es delgada como el papel. James y colaboradores (Circulation, 1996), sugieren que la anomalía de Uhl y la DAVD comparten una patogénesis similar. El diagnóstico diferencial definitivo sólo podría confirmarse mediante la autopsia8,10.
La DAVD se manifiesta generalmente en forma de episodios de TV, con morfología de BRIHH y origen en el VD en adolescentes o adultos jóvenes aparentemente sanos. Las arritmias ventriculares pueden ser asintomáticas y detectarse en un ECG sistemático o pueden causar palpitaciones, síncope o muerte súbita. La edad a la que se produce la primera manifestación oscila entre los 15 y 35 años. Las formas de presentación clínica incluyen palpitaciones, fatiga, dolor torácico atípico, síncope y muerte súbita cardíaca. El trastorno afecta a varones con mayor frecuencia que a mujeres, y suele manifestarse en ellos con una expresión más amplia de la enfermedad. La insuficiencia cardíaca sintomática es una manifestación poco común de la DAVD y la mayor parte de las veces se produce en estadios avanzados de la enfermedad. Los pacientes con antecedentes prolongados de DAVD tienen afectado el ventrículo izquierdo y sufren síntomas clínicos de insuficiencia cardíaca biventricular8,17.
El diagnóstico de miocardiopatía arritmogénica se basa en la presencia de factores estructurales, histológicos, electrocardiográficos, arrítmicos, genéticos y en los antecedentes familiares. Según el Task Force Report publicado por McKenna et al.18 en 1994, los pacientes deben cumplir dos criterios mayores, o un criterio mayor y dos menores, o cuatro criterios menores para que se los considere afectados por una DAVD. Recientemente se ha publicado una nueva modificación de los criterios diagnósticos con la finalidad de aumentar la sensibilidad diagnóstica19.
El ECG de los pacientes con DAVD suele mostrar un ritmo sinusal regular, con una duración del QRS > 110 ms en la derivación V1. Las alteraciones electrocardiográficas incluyen ondas T invertidas en las derivaciones precordiales derechas más allá de V1, sin que haya bloqueo de rama derecha del haz de His y potenciales tardíos ventriculares derechos en forma de «ondas épsilon» en las derivaciones V1-V3. La inversión de la onda T en estas derivaciones representan una característica bien conocida del ECG en la DAVD y, en ausencia de bloque de rama derecha, se ha propuesto que constituye un criterio diagnóstico mayor. Esta variante está presente en un 1-3 % de la población sana de 19 a 45 años de edad, pero se da en el 87 % de los pacientes con DAVD. Las ondas épsilon son potenciales eléctricos de «postexcitación», de pequeña amplitud, que se producen en el segmento ST tras el final del complejo QRS. Estas ondas, que se observan en el 33 % de los pacientes con DAVD, se consideran un criterio diagnóstico mayor19.
Las técnicas de diagnóstico por imagen utilizadas para diagnosticar anomalías morfofuncionales compatibles con una DAVD son: la angiografía convencional, la ecocardiografía, la tomografía computarizada, la angiografía radioisotópica y la resonancia magnética. La angiografía ventricular derecha se ha considerado históricamente la mejor exploración de imagen para el diagnóstico de la DAVD y se ha demostrado que tiene una elevada especificidad (90 %). La ecocardiografía es inocua y constituye un método de primera línea para evaluar a los pacientes en los que se sospecha una DAVD y para la detección sistemática de sus familiares. La resonancia magnética permite diferenciar la grasa del músculo; además, efectuar una evaluación cuantitativa y muy exacta del tamaño y la función del VD. La sensibilidad y la especificidad de la detección de la grasa intramiocárdica del VD mediante resonancia magnética en el diagnóstico de la DAVD, son variables y oscilan entre el 22 y el 100 %. La identificación de la grasa puede ser difícil, ya que el VD es una estructura delgada y las áreas de miocardio afectadas pueden ser muy pequeñas. Además, actualmente es bien sabido que la presencia de grasa en el miocardio del VD puede ser normal20-22. Diferenciar una infiltración adiposa patológica en áreas en que normalmente está presente la grasa epicárdica adyacente, como en el surco aurículo-ventricular y en la parte ántero-apical del VD, puede ser especialmente difícil. También se han descrito zonas aisladas de sustitución grasa en pacientes ancianos, con el uso prolongado de corticoides, en la obesidad, en otras miocardiopatías y en la TV del tracto de salida del VD (TSVD) idiopática. Se ha señalado que demostrar la presencia de tejido fibroso tiene mayor importancia diagnóstica que la observación de grasa sola22,23.
El diagnóstico diferencial principal de la DAVD es el que debe hacerse con la TV del TSVD, la sarcoidosis, la miocardiopatía dilatada idiopática y la miocarditis aislada. Tanto la TV del TSVD como la DAVD se producen en individuos jóvenes aparentemente sanos, y ambas pueden manifestarse por extrasístoles ventriculares o taquicardia ventricular con un BRIHH y un eje inferior. Aunque no es difícil diagnosticar un caso manifiesto de DAVD, la diferenciación de ésta en sus fases iniciales respecto a la taquicardia del TSVD, un trastorno arrítmico generalmente benigno y que no tiene carácter familiar, continúa siendo un verdadero reto clínico.
Los principales factores que determinan una mala evolución son la disfunción grave del VD, la afección del ventrículo izquierdo, el síncope, la edad temprana, el sexo masculino, los antecedentes de parada cardíaca, la TV rápida y mal tolerada con diferentes morfologías, y la incidencia familiar de muertes súbitas juveniles. Los pacientes de muy alto riesgo presentan signos clínicos de insuficiencia cardíaca derecha y pueden tener una disfunción ventricular izquierda, así como antecedentes de TV24.
El objetivo principal de la estrategia terapéutica en la DAVD es la prevención de la muerte súbita cardíaca. Los tres principales tratamientos con que contamos son los fármacos antiarrítmicos, la ablación por catéter y el uso de desfibrilador automático implantable (DAI). Los pacientes con DAVD sin antecedentes de síncope o parada cardíaca, que presentan extrasístoles ventriculares en parejas o salvas cortas, no suelen tener un aumento del riesgo arrítmico y, por consiguiente, no requieren de un tratamiento antiarrítmico específico. En los pacientes con TV mantenida, el tratamiento con fármacos antiarrítmicos tiene como objetivo no solo la supresión de las recurrencias de la TV, sino principalmente la prevención de la muerte súbita cardíaca. El sotalol, a dosis de 320-480 mg/día, ha sido identificado como el fármaco que proporciona mejores resultados, con una tasa general de eficacia de 68 %. La amiodarona, fármaco clase III, ha demostrado eficacia en el tratamiento de las arritmias malignas25,26.
Las indicaciones actuales para la ablación por catéter en pacientes con DAVD son la TV monomórfica bien tolerada, con formas localizadas de la enfermedad y refractariedad a la medicación, o la TV incesante o con descargas frecuentes del DAI. En este último caso, la ablación por catéter puede desempeñar un papel importante como opción de tratamiento paliativo o adyuvante para la reducción o la supresión de la TV26. En la DAVD es el resultado de un circuito de reentrada relacionado con una cicatriz, de manera similar a lo observado después de un infarto de miocardio. La ablación por catéter con mapas de voltaje del VD, mediante el empleo de técnicas de elaboración de mapas convencionales o electroanatómicas, puede aportar unos resultados favorables a corto plazo27.
El tratamiento con DAI mejora el pronóstico a largo plazo y la supervivencia si se aplica a una población de alto riesgo seleccionada y como prevención secundaria. Una complicación frecuente es la debida a la progresión de la atrofia miocárdica y la posterior sustitución por grasa en el lugar de la implantación del electrodo, lo cual da lugar a una pérdida de la función de percepción del electrodo de desfibrilación del VD, que hace necesario sustituirlo. Así pues, la indicación para el tratamiento con DAI en la DAVD debe ponderar los posibles efectos beneficiosos en comparación con el riesgo de complicaciones. Cuando la enfermedad ha progresado a una insuficiencia ventricular derecha o biventricular, se debe aplicar el tratamiento indicado actualmente para la insuficiencia cardíaca, que incluye diuréticos, bloqueadores beta, inhibidores de la enzima de conversión de angiotensina y anticoagulantes. En caso de insuficiencia cardíaca derecha refractaria, el trasplante de corazón puede ser la única alternativa28,29.
La progresión de la enfermedad es incierta e individual para cada paciente y puede ser causa de muerte súbita en jóvenes, o constituir un hallazgo en necropsias de pacientes añosos. La comprensión de su base genética, sus características estructurales y funcionales permitirá en el futuro la búsqueda de nuevas terapias en la prevención, tratamiento y seguimiento de estos pacientes con esta rara enfermedad30.
Conflictos de intereses
Los autores de este trabajo declaran que no existen conflictos de intereses.
Referencias bibliográficas
Frank R, Fontaine G, Mialet G, Sol C, Guiraudon G, Grosgogeat Y. Electrocardiologie de cas de dysplasie ventriculaire droite arythmogene. Arch Mal Coeur Vaiss. 1978;71(9):963-72.
Thiene G, Nava A, Corrado D, Rossi L, Pennelli N. Right ventricular cardiomiopathy and sudden death in young people. N Engl J Med. 1988;318(3):129-33.
Fontaine GH, Guiraudon G, Frank R. Stimulation studies and epicardial mapping in ventricular tachycardia: study of mechanisms and selection for surgery. In: Kulbertus He, ed. Re-entrant arrhythmias: mechanisms and treatment. Baltimore: University Park Press; 1977. p. 334-50.
Anderson EL. Arrhythmogenic right ventricular dysplasia. Am Fam Physician. 2006;73(8):1391-8.
Gallo P, D´Amati G, Pelliccia F. Pathologic evidence of extensive left ventricular involvement in arrhythmogenic right ventricular cardiomyopathy. Hum Pathol. 1992;23(8):948-52.
Thiene G, Basso C. Arrhythmogenic right ventricular cardiomyopathy: an update. CardiovascPathol. 2001;10(3):109-17.
Marcus FI, Fontaine GH, Guiraudon G, Frank R, Laurenceau JL, Malergue C, et al. Right ventricular dysplasia: a report of 24 adult cases. Circulation. 1982;65(2):384-98.
Thiene G, Corrado D, Basso C. Arrhythmogenic right ventricular cardiomiopathy/dysplasia. Orphanet J Rare Dis. 2007;2:45.
Kayser H, Van der Wall EE, PleinS,Bloomer TN, de Roos A. Diagnosis of arrhythmogenic right ventricular dysplasia: a review. Radiographics. 2002; 22(3):639-48.
Morales Pérez LM, Jerez Castro AM, San Román García E. Displasia arritmogénica del ventrículo derecho o enfermedad de UHL. RevFedArgCardiol. 2012;41(1):59-60.
Gaffney FA, Nicod P, Lin JC, Rude RE. Noninvasive recognition of the parchment right ventricle (Uhl´s anomaly arrhythmogenic right ventricular dysplasia) syndrome. ClinCardiol. 1983;6(5):235-42.
Daliento L, Rizzoli G, Thiene G, Nava A, Rinuncini M, Chioin R, et al. Diagnostic accuracy of right ventriculography in arrhythmogenic right ventricular cardiomyopathy. Am J Cardiol. 1990;66(7):741-5.
Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the Task Force Criteria. Eur Heart J. 2010;31(7):806-14.
Rampazzo A, Nava A, Malacrida S, Beffagna G, Bauce B, Rossi V, et al. Mutation in human desmoplakin domain binding to plakoglobin causes a dominant form of arrhythmogenic right ventricular cardiomyopathy. Am J HumGenet. 2002;71(5):1200-6.
Gerull B, Heuser A, Wichter T, Paul M, Basson CT, McDermott DA, et al. Mutations in the desmosomal protein plakophilin 2 are common in arrhythmogenic right ventricular cardiomiopathy. Nat Genet. 2004;36(11):1162-4.
Capulzini L, Brugada P, Brugada J, Brugada R. Arritmias y enfermedades del corazón derecho: de las bases genéticas a la clínica.Rev EspCardiol. 2010;63(8):964-5.
Corrado D, Basso C, Thiene G, McKenna WJ, Davies MJ, Fontaliran F, et al. Spectrum of clinicophatologic manifestations of arrhythmogenic right ventricular cardiomiopathy/dysplasia: a multicenter study. J A CollCardiol. 1997;30(6):1521-30.
McKenna WJ, Thiene G, Nava A, Fontaliran F, Blomstrom−Lundqvist C, Fontaine G, et al. Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Task Force of the Working Group Myocardial and Pericardial Disease of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology. Br Heart J. 1994;71(3):215-8.
Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke D, et al. Diagnosis of arrythmogenic right ventricular cardiomyopathy/dysplasia. Proposed modification of the Task Force criteria. Circulation. 2010;121(13):1533-41.
Daliento L, Rizzoli G, Thiene G, Nava A, Rinuncini M, Chioin R, et al. Diagnostic accuracy of right ventriculography in arrhythmogenic right ventricular cardiomyopathy. Am J Cardiol. 1990;66:741-5.
Van der Wall EE, Kayser HW, Bootsma MM, De RoosA, Schalij MJ. Arrhythmogenic right ventricular dysplasia: MRI findings. Herz. 2000;25(4):356-64.
Tandri H, Bomma C, Calkins H, Bluemke DA. Magnetic resonance and computed tomography imaging of arrhythmogenic right ventricular dysplasia. J MagnReson Imaging. 2004;19(6):848-58.
Burke AP, Farb A, Tashko G, Virmani R. Arrhythmogenic right ventricular cardiomyopathy and fatty replacement of the right ventricular myocardium: are they different diseases? Circulation. 1998;97(16):1571-80.
Hulot JS, Jouven X, Empana JP, Frank R, Fontaine G. Natural history and risk stratification of arrhythmogenic right ventricular dysplasia/cardiomiopathy. Circulation. 2004;110:1879-84.
Wichter T, Borggrefe M, Haverkamp W, Chen X, Breithardt G. Efficacy of antiarrhythmic drugs in patients with arrhythmogenic right ventricular disease: results in patients with inducible and noninducible ventricular tachycardia. Circulation. 1992;86(1):29-37.
Marcus G, Glidden D, Polonsky B, Zareba W, Smith L, Cannom D, et al. Efficacy of antiarrhythmic drugs in arrhythmogenic right ventricular cardiomiopathy. J Am CollCardiol. 2009;54(7):609-15.
Arruda M, Armaganijan TF, Di Biase L, Patel D, Natale A. Catheter ablation of ventricular tachycardia in arrhythmogenic right ventricular dysplsia. J IntervCardElectrophysiol. 2009;25(2):129-33.
Wichter T, Brethardt G. Implantable cardioverter−defibrillator therapy in arrhythmogenic right ventricular cardiomiopathy: a role for genotyping in decision−making? J Am CollCardiol. 2005;45(3):409-11.
Hodgkinson KA, Parfrey PS, Bassett AS, Kupprion C, Drenckhahn J, Norman MW, et al. The impact of implantable cardioverter defibrillator therapy on survival in autosomal-dominant arrhythmogenic right ventricular cardiomiopathy (ARVD5). J Am CollCardiol. 2005;45(3):400-8.
Turrini P, Basso C, Daliento L, Nava A, Thiene G, et al. Is arrhythmogenic right ventricular cardiomyopathy a pediatric problem too? Images PaediatrCardiol. 2000;3(1):18-37.