



CorSalud 2014;6(Supl 1)
CONFERENCIA
DETERMINANTES GENÉTICAS DE LA MUERTE SÚBITA CARDIOVASCULAR
MSc. Dra. Norma E. de León Ojeda
______________
Hospital Pediátrico "William Soler". La Habana, Cuba.
Correspondencia: NE de León Ojeda. Cardiocentro Pediátrico William Soler. Ave. 100 y Perla, Altahabana. Boyeros, CP 10800. La Habana, Cuba. Correo electrónico: norma.deleon@infomed.sld.cu
Versión impresa de la conferencia impartida en el marco del I Simposio Cubano de Muerte Súbita Cardiovascular celebrado en La Habana, Cuba, del 7-9 de noviembre de 2013.
Resumen
Introducción: La muerte súbita cardiovascular (MSC) ocurre de manera inesperada en las primeras seis horas del inicio de los síntomas. Entre las causas más comunes están la cardiopatía isquémica (80 %) y las enfermedades genéticas con alteraciones estructurales o funcionales. La genética molecular ha identificado sustratos condicionantes de muerte súbita cardiovascular que siguen patrones de herencia mendeliana como las canalopatías y las miocardiopatías hereditarias, enfermedades heterogéneas clínica y molecularmente, con igual fenotipo clínico o electrocardiográfico. Objetivo: Actualizar los conocimientos sobre esta temática desde la óptica de la genética clínica y los resultados de estudios moleculares existentes, así como aplicarlos a casos clínicos aislados y familiares evaluados clínicamente. Método: Se realizó una búsqueda actualizada sobre muerte súbita cardiovascular relacionada con los factores genéticos causales o condicionales y se revisaron las historias clínicas de pacientes con condiciones genéticas relacionadas en el Servicio de Genética del Hospital William Soler. Resultados: Se actualiza la temática desde la arista molecular en su relación clínica y se presentan las experiencias con la evaluación y conducta clínica de una familia cubana con síndrome de QT prolongado, con heterogeneidad clínica, electrocardiográfica y estudio molecular no concluyente. Se relacionan otros casos con muerte súbita cardiovascular en un lactante con sindactilia y una transicional con Síndrome de Costello. Conclusiones: Los genes de susceptibilidad y predisposición para las causas cardiovasculares de muerte súbita son muy heterogéneos y dependientes de modificaciones epigenéticas en su expresión, por lo que la muerte súbita cardiovascular es un desafío contemporáneo para la cardiología en que la genética clínica y la molecular pueden aportar elementos definitorios para su prevención y tratamiento.
Palabras clave: Muerte súbita, Defectos cardiovasculares congénitos, Canalopatías
Genetic determinants of sudden cardiac death
Abstract
Introduction: Sudden cardiac death occurs unexpectedly in the first six hours after the onset of symptoms. The most common causes include ischemic heart disease (80 %) and genetic diseases with structural or functional abnormalities. Molecular genetics has identified substrates that are determinants of cardiovascular sudden death, which follow Mendelian inheritance patterns such as hereditary channelopathies and cardiomyopathies, clinical and molecularly heterogeneous diseases, with the same clinical or electrocardiographic phenotype. Objective: To update knowledge on this subject from the perspective of clinical genetics and the results of existing molecular studies, as well as applying them to isolated clinical cases and family cases that have been clinically assessed. Method: An updating search on sudden cardiac death was conducted. It was focused on causal or determining genetic factors. The clinical records of patients with associated genetic conditions were reviewed at the Genetics Department of the William Soler Hospital.Results: The topic is updated from the molecular perspective and its clinical relationship. The experiences with the assessment and clinical conduct in a Cuban family with long QT syndrome, clinical and electrocardiographic heterogeneity and inconclusive molecular study are reported. Other cases with sudden cardiac death are reported, in an infant with syndactyly, and a transitional one with Costello syndrome. Conclusions: The genes of susceptibility and predisposition to cardiovascular causes of sudden death are very heterogeneous and dependent on epigenetic modifications in its expression. Therefore, sudden cardiac death is currently a challenge to cardiology, in which clinical and molecular genetics may contribute defining elements to its prevention and treatment.
Key words: Sudden death, Congenital cardiovascular defects, Channelopathies
Introducción
La muerte súbita (MS) probablemente sea el desafío más importante de la cardiología moderna por la frecuencia de presentación y las alternativas preventivas respecto a la causas. Se considera MS la que ocurre de manera inesperada dentro de las primeras seis horas desde el inicio de los síntomas o si se produce en ausencia de testigos cuando el fallecido ha sido visto en buenas condiciones menos de 24 h antes de hallarlo muerto1,2.
La MS de origen cardíaco (MSC) representa más del 90 % de todos los casos y puede ser de tipo arrítmico, mucho más frecuente, o por fallo cardíaco. En la de tipo arrítmico la pérdida de conciencia y la falta de pulso arterial se presentan en ausencia de colapso circulatorio, mientras que el fallo cardíaco se produce progresivamente y conduce al colapso circulatorio antes de que se presente el paro cardíaco3.
La MSC de tipo arrítmico puede estar precedida de episodios sincopales, con una pérdida transitoria de conciencia, que cursa con recuperación espontánea y sin secuelas debido a que la hipoperfusión cerebral es general y transitoria. Sólo del 6-30 % de los síncopes son de causa cardíaca, que a veces es la primera manifestación de su enfermedad y puede ser un marcador de mal pronóstico, con riesgo de MS4.
La MSC es rara en las primeras décadas de la vida, aunque puede verse en lactantes relacionada con trastornos de la repolarización, alteraciones del sistema nervioso autónomo e incremento del tono vagal, durante la actividad deportiva, y en presencia de cardiopatías de origen genético. Antes de los 35-40 años es relativamente frecuente la asociación a miocardiopatía hipertrófica y miocarditis subclínicas, sobre todo en deportistas jóvenes. En menor frecuencia pueden asociarse la preexcitación ventricular tipo Wolff-Parkinson-White, displasia arritmogénica de ventrículo derecho (DAVD), o algunas valvulopatías y anomalías congénitas de las coronarias1.
La incidencia de MS aumenta significativamente a partir de los 35-40 años, y es alta en la fase aguda o crónica del infarto de miocardio. La cardiopatía isquémica es causa de la MS aproximadamente en el 80 % de los casos, pero entre 3-15 % es causada por enfermedades de origen genético. Se estima que el 15 % de las MSC tienen alteraciones estructurales (miocardiopatías, DAVD) y el 3-5 % son por alteraciones funcionales (canalopatías), que explican muchos de los casos de MS en la juventud relacionados o no con el esfuerzo, pero en individuos sin cardiopatía isquémica1.
Los avances en la genética molecular han permitido identificar sustratos genéticos subyacentes a la patogenia de condiciones potencialmente mortales que siguen patrones de herencia mendeliana, como las canalopatías y las miocardiopatías hereditarias, enfermedades heterogéneas clínica y molecularmente, causadas por muchas mutaciones (heterogeneidad alélica) en varios genes (heterogeneidad no alélica), que originan el mismo fenotipo clínico o electrocardiográfico5.
FACTORES GENÉTICOS
Resulta categórico hablar sobre "determinantes" de la MSC cuando los factores genéticos están relacionados causalmente con genes de susceptibilidad o genes predisponentes con efectos mayores, que condicionan al miocardio vulnerable, y ante la presencia de factores desencadenantes se expresan clínicamente en fenotipos cardíacos o electrofisiológicos. Esto supone que la herencia multifactorial con una base genética más el ambiente (micro o macroambiente / endógeno o exógeno), es responsable de los efectos clínicos en esta afección, aunque estén descritos patrones de herencia autosómicos dominantes o recesivos. De esta interacción genoma-ambiente depende la penetrancia reducida y la expresividad variable que muestran muchas de estas enfermedades con patrones de herencia bien definidos. La búsqueda de elementos genómicos y la correlación fenotipo-genotipo como elementos pronósticos, constituyen claves para el diagnóstico y la conducta a seguir en muchos casos. Siendo así podríamos definir factores condicionantes (mutaciones) y detonantes (ambientales) de la MSC.
Los tres factores que con más frecuencia hacen al miocardio vulnerable son a) la isquemia, b) la disfunción del ventrículo izquierdo y c) la predisposición/susceptibilidad genética. Los dos primeros forman parte del endofenotipo, también de causa genética multifactorial y el tercero probablemente tenga un papel protagonista.
Las categorías de susceptibilidad y predisposición son indistintamente utilizadas, pero no tienen el mismo significado semántico y en términos genéticos pueden diferir. Según el diccionario susceptible se refiere a lo que es capaz de recibir una modificación o impresión; y predispuesto, a lo dispuesto de manera anticipada hacia algo6. Pudiera establecerse una diferencia en el sentido genético de ambos términos relacionada con la interacción del genoma con los factores ambientales y así, la predisposición podría referirse a la existencia de genes con una contribución mayor a la aparición de un fenotipo que no necesariamente depende de la interacción con un ambiente desfavorable; y susceptibilidad, a la existencia de genes que solo en presencia de un ambiente desfavorable se expresan en el fenotipo, son causa necesaria pero no suficiente para la aparición de una enfermedad genética.
Los genes de susceptibilidad y predisposición pueden ser responsables de los sustratos morfofisiológicos, como la hipertrofia ventricular izquierda y la inestabilidad eléctrica (arritmias ventriculares, disfunción del sistema nervioso autónomo). Estas condicionantes pueden estar presentes meses o años sin que ocurra la MS en ausencia de factores desencadenantes. Los agentes ambientales que actúan como desencadenantes, son el estrés físico o psíquico, las alteraciones iónicas o metabólicas, la administración de fármacos con efecto arritmogénico que provocan torsades de pointes, deprimen la contractilidad, interfieren en la isquemia y favorecen la aparición de MS3.
De esta interacción genoma-ambiente depende el fenotipo clínico y electrofisiológico en la MSC, así como la expresividad variable y penetrancia reducida, que tienen una expresión tan individual y dependen del endofenotipo y de la interacción entre productos génicos y agentes ambientales.
Grupos de enfermedades o condiciones clínicas que causan MSC1:
Cardiopatía isquémica e insuficiencia cardíaca.
Alteraciones electrofisiológicas por arritmias o alteraciones del sistema de conducción.
Canalopatías: son enfermedades sin afección estructural aparente o sólo mínima. Ejemplos: Síndromes del QT largo, QT corto, Síndrome de Brugada y la taquidarcia ventricular (TV) catecolaminérgica, la fibrilación auricular familiar y las arritmias ventriculares malignas, como la TV helicoidal familiar y la fibrilación ventricular, considerada hasta ahora como idiopática, e incluso, probablemente algunos casos de bradicardia sinusal de origen genético y el síndrome de Wolff-Parkinson-White.
Taquiarritmias supraventriculares: son los ritmos rápidos que se mantienen por alguna estructura por encima de la bifurcación del haz de His, necesaria para su mantenimiento.
Alteración del sistema de conducción (enfermedad de Lenegre).
Miocardiopatías: las miocardiopatías debidas a alteraciones de las proteínas cardíacas.
Miocardiopatías hipertrófica y dilatada.
DAVD.
Miocardiopatía no compactada.
Cardiopatías congénitas estructurales: anomalías congénitas de las coronarias, valvulopatías, tetralogía de Fallot operada, entre otras.
MSC sin causa aparente que incluye el Síndrome de muerte súbita del lactante.
FACTORES GENÉTICOS SUBYACENTES EN LAS ENFERMEDADES QUE CAUSAN MSC
1. Cardiopatía isquémica e insuficiencia cardíaca
Esta enfermedad vascular de herencia multifactorial puede asociarse a factores genéticos que desencadenan taquicardia o fibrilación ventriculares durante los síndromes coronarios agudos con elevación del ST. Hay variantes genéticas relacionadas con riesgo para la fibrilación ventricular en el infarto agudo de miocardio.
Locus en el cromosoma 21, el alelo raro del polimorfismo rs2824292 [índice de probabilidad (OR, en inglés) = 1,49; intervalo de confianza de 95 %; 1,14-1,95] y el gen más cercano a este locus es CXADR, que codifica un receptor viral implicado previamente en miocarditis y miocardiopatía dilatada, que modula la conducción del impulso eléctrico cardíaco.
El alelo raro del polimorfismo rs3864180 localizado en el gen GPC5 se asocia con menos riesgo de MS para los pacientes con cardiopatía isquémica [índice de riesgo (hazard ratio, en inglés) = 0,85; intervalo de confianza de 95 %; 0,74-0,98). La presencia de este alelo también se asoció a una menor duración del intervalo QT.
El alelo raro del polimorfismo rs4665058 se asocia con mayor riesgo de MS para los pacientes con cardiopatía isquémica crónica (OR = 1,91)1.
Algunos autores han estudiado del rol de la mutación SCN5A G400A en el desarrollo de tormentas arrítmicas durante la isquemia aguda, así como el fenotipo 4G/4G del gen PAI-1 que codifica para el activador e inhibidor del plasminógeno, que condiciona arritmias ventriculares malignas y un estado protrombótico y antifibrinolítico en pacientes con enfermedad coronaria7. Como biomarcador se estudia la actividad de la proteína quinasa calmodulina-dependiente (CaMKII), relacionada con insuficiencia cardíaca, hipertrofia y arritmias8.
2. Alteraciones electrofisiológicas o del sistema de conducción
Canalopatías: En este grupo se incluyen síndromes genéticos causados por mutaciones en proteínas que funcionan como canales, que tienen patrones de herencia mendelianos clásicos pero cuya expresividad es variable clínica y electrofisiológicamente por causas aún en estudio.
2.1. Síndrome de QT largo (SQTL)
El SQTL explica un 20 % de las muertes súbitas en jóvenes con necropsia negativa y un 10 % de las muertes por síndrome de muerte súbita infantil. Solo el 5-10 % de los casos con SQTL son causados por mutaciones de novo y el resto son heredadas. Se conocen cuatro formas hereditarias de este síndrome, el síndrome de Romano-Ward, el de Andersen-Tawil, el de Timothy y el de Jervell-Lange-Nielsen. El de Romano-Ward, con prevalencia de 1/5.000 individuos, representa el 85 % de todos los casos de SQTL. Tiene un patrón de transmisión autosómico dominante con 12 genes mayores identificados en su etiopatogenia, que originan un fenotipo de SQTL clásico no sindrómico, y otros trastornos multisistémicos, como el Síndrome por anquirina B, el de Anderson-Tawil y el de Timothy, anteriormente LQT4, LQT7 y LQT8, respectivamente3,5,9.
El fenotipo más grave del síndrome de Jervell-Lange-Nielsen tiene el QT prolongado acompañado de hipoacusia neurosensitiva y es de herencia autosómica recesiva, pero con mutaciones en los genes mayores del SQTL en homocigosis, ejemplo de relatividad en los patrones de herencia respecto a los efectos de la interacción de los productos génicos en su expresión clínica. Las combinaciones de mutaciones en KCNQ1 (OMIM 607542) y KCNE1 (OMIM 176261), como heterocigóticos compuestos, causan este síndrome genético con trastornos en la homeostasis de la endolinfa y sordera, además de arritmias malignas10.
Esta condición genética puede expresarse clínicamente con arritmias cardíacas y aumento del intervalo QT en el electrocardiograma. Los síntomas pueden aparecer en situaciones de estrés o por ingestión de medicamentos, aunque muchos de estos casos pueden permanecer asintomáticos durante toda su vida. El SQTL tiene una penetrancia de un 25-90 %, por lo que existe la posibilidad de padecer la enfermedad con un electrocardiograma normal. Aunque la causa de este síndrome no sólo implica a las canalopatías, recientemente se ha señalado la posible utilidad del estudio de los genes implicados en SQTL9.
Aproximadamente un 75 % de los SQTL se deben a mutaciones de tres genes considerados como mayores –KCNQ1 (LQT1), KCNH2 (LQT2) y SCN5A (LQT3)– que codifican los poros del canal iónico de potasio (Kv7.1 y Kv11.1) o de sodio (Nav1.5), y modulan los potenciales de acción de los miocitos ventriculares. Los siete genes menores de susceptibilidad al SQTL explican menos del 5 % de los casos como, los genes KCNJ2 del síndrome de Andersen-Tawil; CACNA1C del síndrome de Timothy, KCNQ1/KCNE1 del síndrome de Jervell-Lange-Nielsen y ANK2, que codifica para la anquirina B, la primera proteína que no está implicada con los canales iónicos3,5,10.
En cuanto a la correlación fenotipo-genotipo del LQT1 se presentan los episodios durante el ejercicio y las ondas T son de base ancha. En LQT2 los episodios son más frecuentes durante el reposo o el sueño, el estímulo auditivo y el puerperio; en este caso las ondas T son de baja amplitud con una escotadura, ó bifásicas. El LQT3 tiene episodios durante el reposo o el sueño, con un segmento isoeléctrico largo seguido de una onda T de base estrecha. Estos elementos no son predictivos, pero orientan al elegir el gen por el que se comenzará el estudio molecular y de este resultado dependerán las acciones terapéuticas con betabloqueadores, que brindan protección máxima al LQT1, moderada al LQT2 e insuficiente al LQT3, por lo que necesita asociarse a mexiletina, flecainida o ranolazina5.
Las mutaciones de cambio de sentido en regiones transmembranales del poro duplican el riesgo de episodios cardíacos más que las mutaciones localizadas en el extremo carboxílico del canal de potasio Kv7.1. Las mutaciones que causan mayor pérdida de función (dominante negativo) duplican el riesgo clínico sobre las haploinsuficiencias. Un 25-30 % de los pacientes con SQTL permanecen sin diagnóstico genético a pesar de la secuenciación completa de todos los genes descritos5,9.
Sin el estudio genético combinado con pruebas farmacológicas se identificaría menos de la mitad de los portadores en familiares asintomáticos, pues en más del 70 % de los casos no cumplen los criterios diagnósticos. La penetrancia y expresividad en portadores de una mutación potencialmente patogénica, es baja; sin embargo, no a todos ellos se los puede considerar pacientes por el hecho de portar una mutación genética, ya que muchos nunca expresan el fenotipo y otros sólo lo expresan en situaciones que alteren la repolarización, como toma de fármacos o trastornos iónicos (portadores asintomáticos). Discernir qué portadores genéticos expresarán el fenotipo y cuáles no, resulta difícil, y en ese sentido las pruebas farmacológicas pueden ser de apoyo11.
Un ejemplo de heterogeneidad clínica en esta enfermedad son las mutaciones del canal de sodio que codifica el gen SCN5A y causan múltiples síndromes arritmogénicos, como el LQT3, el de Brugada, trastornos de conducción, enfermedad del nodo, arritmias auriculares y otras. La mutación SCN5A 1795insD es un ejemplo clave para ilustrar este efecto, pues los pacientes que la presentan pueden expresar clínicamente bradicardia, defectos de conducción, LQT3 o Brugada. En estos casos se han realizado estudios de función génica para explicar esta variabilidad fenotípica y los del promotor del gen han develado asociación de dos SNP (siglas en inglés de polimorfismos de nucleótidos simples) a la intensidad del fenotipo con independencia de los perfiles de metilación de genes relacionados; por lo cual las variantes específicas del promotor del gen, inciden en el riesgo de un fenotipo más grave. También existen elementos represores/promotores tejido-específicos y variantes de empalme alternativo que condicionan la complejidad de la expresión clínica12.
2.2. Síndrome de Brugada (SB)
El síndrome del bloqueo de rama derecha, elevación persistente del segmento ST y MS, más conocido hoy en día como síndrome de Brugada, fue descrito en 1992 como un nuevo síndrome clínico-electrocardiográfico causante de arritmias ventriculares y MS, en pacientes sin cardiopatía estructural evidente. Hoy se clasifica como una enfermedad eléctrica primaria o canalopatía cardíaca13-15.
Esta enfermedad se caracteriza por episodios de TV polimórfica rápida con episodios sincopales o MS, generalmente durante el sueño. Su herencia es autosómica dominante, relacionada con el gen SCN5 y presenta mayor incidencia en hombres (8:1), principalmente adultos jóvenes (30-40 años de edad) sin evidencias de cardiopatía estructural previa. Se estima que entre 4-12 % de las MS y hasta un 20 % de las MSC en individuos que no presentan cardiopatías estructurales, se dan como consecuencia de este síndrome. Su prevalencia es difícil de estimar por lo difícil de identificar sus formas de presentación, pero se calcula en 5/10.000 individuos3,5,13.
Electrocardiográficamente se expresa con elevación del segmento ST de morfología descendente (2 mm) seguida de una onda T negativa registrada en las derivaciones precordiales derechas (V1-V3, patrón electrocardiográfico de SB tipo 1), y un aumento de la predisposición a la MS. Hay descritos otros dos patrones electrocardiográficos, tipos 2 y 3, que no se consideran diagnósticos de SB. Solo el patrón tipo I lo es, y las observaciones fisiopatológicas indican que los trastornos eléctricos relacionados con el SB se sitúan principalmente en el ventrículo derecho, en especial en su tracto de salida (TSVD). La MSC se produce por una TV polimórfica o fibrilación ventricular5,13-15.
Aunque un 70-80 % de los casos de SB continúan sin esclarecerse genéticamente, se han identificado 15 genes de susceptibilidad. Las mutaciones de pérdida de función en la subunidad A del canal de sodio cardíaco, codificado en SCN5A, constituyen el sustrato genético más frecuente del SB y explican un 15-30 % de los casos del trastorno. Aparte de la disfunción del canal de sodio, las mutaciones que afectan al canal de calcio de tipo L en las subunidades A1 (CACNA1C), B2 (CACNB2B) y A2d (CACNA2D1) pueden causar un 10-15 % de los casos de SB, especialmente cuando se acompañan de intervalos QT breves. También el gen que codifica la glicerol-3-fosfato deshidrogenasa 1 (enzima que regula el tráfico por el canal de sodio cardíaco hacia la superficie celular y reduce las corrientes de entrada de sodio en aproximadamente un 50 %), y las mutaciones del gen KCNE3, que codifica una subunidad reguladora de la corriente Ito de potasio, producen un fenotipo de SB5,13.
Existe gran heterogeneidad clínica en las canalopatías que se corresponde con la expresividad variable, pero no pueden olvidarse los efectos epigenéticos ambientales y de la interacción de los productos génicos (epistasia), que han dado lugar a los llamados «síndromes solapados». Se ha informado concomitancia de SB y enfermedad de conducción cardíaca (enfermedad de Lev-Lenegre, síndrome del nódulo sinusal enfermo); entre los fenotipos LQT3 y SB, y la asociación de la fibrilación auricular con canalopatías de sodio conocidas, como el SB, la enfermedad de conducción cardíaca progresiva, los síndromes de QT corto y el LQT3. El síncope de mecanismo neurógeno se ha asociado recientemente al SB, pero no se conocen todavía sus repercusiones en cuanto al pronóstico y la estratificación del riesgo13.
El papel principal de las pruebas genéticas es confirmar el diagnóstico en el paciente y en los familiares inmediatos, para diferenciar a los que requieren vigilancia clínica continua y medidas preventivas como evitar algunos fármacos, de aquellos que se puede considerar sin afección clínica, electrocardiográfica y genética. Aunque puede usarse quinidina como opción de tratamiento farmacológico, el único tratamiento que ha demostrado la prevención de la MS en pacientes con SB sintomáticos es el desfibrilador automático implantable (DAI)12,13.
2.3. Taquicardia ventricular polimórfica catecolaminérgica (TVPC)
Esta enfermedad se presenta en corazones estructuralmente normales, se desencadena ante episodios de liberación de catecolaminas (noradrenalina y adrenalina) en situaciones de estrés físico o emocional. Su prevalencia, actualmente es desconocida, se estima en 1/2.000 individuos y el 30 % tienen antecedentes familiares de MS o síncope, y se considera causa de un 15 % de las MS inexplicadas en sujetos jóvenes con necropsia negativa3,5.
Los síntomas aparecen entre los 5 y 10 años; sin embargo, los casos de MS son raros a esta edad. La tasa de mortalidad aumenta a 30-50 % al llegar a los 35 años, lo que se ha relacionado con factores, como el sexo masculino, el consumo de tabaco, la obesidad, la diabetes, la hipertensión arterial y los niveles elevados de colesterol LDL3,5.
En su mayoría el patrón de herencia es autosómico dominante (OMIM 614021), hasta un 60 % de las familias muestran mutaciones en el gen que codifica para el receptor de la rianodina con alteraciones en la liberación de calcio del retículo sarcoplásmico, con retraso en las despolarizaciones y arritmias. También existe un patrón de herencia autosómico recesivo, debido a mutaciones en el gen CASQ2, el cual codifica para la proteína calciquestrina 2 (OMIM 114251). Los pacientes con presencia de una TV bidireccional con el ejercicio tienen mutaciones de KCNJ2, gen relacionado también con el Síndrome de Andersen-Tawil tipo 1, que no requiere tratamiento profiláctico agresivo, es decir, DAI. Las pruebas genéticas pueden aportar una diferenciación diagnóstica clara entre la TVPC y el SQTL tipo LQT1 oculto, y entre la TVPC y el Anderson-Tawil tipo 13,5,10.
2.4. Taquicardia ventricular de tracto de salida de ventrículo derecho
La TV-TSVD se produce más frecuentemente entre los 30-50 años, en las mujeres y en un 70-80 % se origina realmente en esta zona, pero también puede originarse en el tabique, el tracto de salida ventricular izquierdo, la arteria pulmonar, el seno de Valsalva aórtico, el área próxima al haz de His y la superficie epicárdica. Es monomórfica y generalmente, no es de carácter familiar. La morfología característica de la TV-TSVD es la de una taquicardia con QRS ancho, con morfología de bloqueo de rama izquierda del Haz de His, y un eje inferior con dos formas fenotípicas: la TV sostenida inducida por el esfuerzo o la tensión emocional y la TV monomórfica repetitiva no sostenida que aparece en reposo. Ambas formas se caracterizan por la sensibilidad a la adenosina y muestran un curso benigno, por lo que la arritmia no es expresión de una miocardiopatía oculta. En estos casos hay sobrecarga de calcio intracelular que potencia la función del intercambiador de Na+/Ca++, con aumento de la corriente de entrada y retraso en la posdespolarización. La adenosina reduce el AMPc, crucial en la regulación del calcio intracelular. Esta arritmia puede interrumpirse de forma aguda con maniobras vagales, masaje del seno carotídeo, adenosina o verapamilo intravenoso. Por la tasa de éxitos a largo plazo de más del 90 % y las escasas complicaciones (< 1 %), la ablación por catéter resulta el tratamiento de primera línea en pacientes con TV-TSVD13,17.
Se ha identificado una mutación puntual en el codón 200 del gen GNAI2 (OMIM 139360), con cambio de fenilalanina por leucina, alteración encontrada solamente en el segmento de origen de la arritmia, evidencia a favor de la presencia de mosaicismo somático como causa de esta enfermedad clínico-electrocardiográfica10.
3. Miocardiopatías
Pueden ser aisladas no sindrómicas o estar asociadas a síndromes genéticos o condiciones metabólicas18.
3.1. Miocardiopatía hipertrófica (MCPH)
Esta enfermedad hereditaria, principal causa de MS en los adultos menores de 50 años, se caracteriza por el engrosamiento importante del músculo cardíaco, principalmente del ventrículo izquierdo. Esta miocardiopatía con prevalencia de 1/500 individuos, constituye una de las enfermedades genéticas más frecuentes. Su herencia es autosómica dominante y se han identificado 27 genes en su etiopatogenia, de los cuales 9 codifican proteínas sarcoméricas. Entre ellos se encuentran los MYL2, MYL3, ACTC, TPM1, TNNT2, TNNI3, TTN (PRKAG2), MYH7 y MYBPC3, estos dos últimos los más frecuentemente mutados2,9,18.
Las pruebas moleculares de 9 genes en los casos esporádicos o familiares identifican el 40-60 % de las mutaciones y el 15-30 % son del gen MYH7 que codifica para la cadena pesada de la betamiosina y otro 25 % del MYBPC3, proteína transportadora de la miosina C cardíaca20-22.
En un estudio reciente en 171 pacientes con MCPH de los 5 genes más frecuentemente implicados en la enfermedad (MYH7, MYBPC3, TPM1, TNNI3 y TNNT2), se identificó la mutación causal en 82 pacientes (48 %). Luego se estudiaron 228 familiares y se constató la mutación en 106 de ellos (46,5%)21.
Los estudios genéticos de la MCPH contribuyen principalmente a proporcionar la confirmación diagnóstica, y sólo tienen un papel menor en cuanto al pronóstico y las repercusiones terapéuticas. Las pruebas genéticas para esta miocardiopatía pueden ser esenciales en la diferenciación entre una MCPH verdadera y una hipertrofia por adaptación al ejercicio u otros trastornos genéticos con hipertrofia miocárdica, como el Síndrome de Fabry, las glucogenosis, las enfermedades lisosomales, mitocondriales y los síndromes de Noonan y LEOPARD*, para los que existen otros tratamientos que difieren de los de la MCPH. Sin embargo, una prueba genética negativa no descarta la MCPH, además de que los tratamientos profilácticos aportan la posibilidad de prevenir o retardar el inicio de la hipertrofia en los pacientes con genotipo positivo pero sin evitarla5,10,19,23.
3.2. Miocardiopatía dilatada (MCPD)
Esta miocardiopatía es una de las causas de MS y de transplantes cardíacos en niños y adultos. Su prevalencia se estima en 1/3.000 individuos y se caracteriza por la dilatación anormal con disfunción sistólica del ventrículo izquierdo3.
De los estudios realizados en pacientes con miocardiopatía dilatada, se han identificado mutaciones en 30 genes. La mayoría de ellas son de herencia autosómica dominante con mutaciones en los genes ACTC, TPM1, MYH7, TNNT2, TNNI3, MYBPC-3, TTN, TCAP, DES, DMD, VCL, SGSD, ACTN2 y, LMNA/C, muchos de ellos codifican proteínas sarcoméricas, citoesqueléticas, miofilamentos y canales iónicos que también causan MCPH. Las relacionadas con la laminina y las desminas son las más relacionadas con la MSC. En otros casos la transmisión familiar es recesiva ligada al X y mitocondrial, con herencia nuclear o mitocondrial3,10.
La interpretación de los resultados de los exámenes genéticos para la MCPD y la DAVD resulta difícil, pues no está clara su repercusión clínica5. El 48 % de los casos con mutaciones génicas para la MCPD son asintomáticos.
La cardiopatía isquémica y las miocardiopatías del ventrículo izquierdo han sido clásicamente las principales causas de arritmias ventriculares y MSC; sin embargo, las arritmias originadas en el ventrículo derecho suelen afectar a pacientes de menor edad y conducir a la MSC. La fisiopatología de esas arritmias no se ha aclarado por completo, tiene diferentes interpretaciones y la genética contribuye a explicar muchos aspectos patogénicos, diagnósticos y pronósticos en estas arritmias. Uno de los mecanismos está relacionado con la disfunción del intercambio de calcio en el retículo sarcoplásmico que da lugar a las arritmias ventriculares en esta cardiopatía7,13,24.
3.3. Displasia arritmogénica del ventrículo derecho
Es una enfermedad del corazón de herencia autosómica dominante, caracterizada por arritmias ventriculares y anomalías estructurales del ventrículo derecho con pérdida progresiva de miocitos reemplazados por tejido fibroadiposo. Actualmente se considera que la disfunción desmosómica es la vía final común en la patogenia de la DAVD. Aunque afecta al ventrículo derecho, también puede comprometer al izquierdo3,13.
Su prevalencia se estima en 1/2.000 a 1/10.000 individuos y el 80 % de los casos afectados son menores de 40 años. La DAVD explica hasta un 5-20 % de la MSC, que puede ser la primera manifestación de la enfermedad sobre todo en varones menores de 30 años por lo difícil de su diagnóstico clínico. Debe sospecharse una DAVD en todos los pacientes jóvenes con un corazón aparentemente normal que sufren un síncope, TV o parada cardíaca y también en deportistas con síntomas, como arritmias, palpitaciones o síncope3,13.
La DAVD es hereditaria, en un 30-50 % de los casos autosómica dominante, con expresividad variable y penetrancia reducida, además de una expresión fenotípica polimórfica. Se han identificado mutaciones en más de 12 genes en la DAVD no sindrómica, muchos de ellos codifican proteínas del desmosoma como la desmocolina 2, desmogleína 2, desmoplakina, factor de crecimiento fibroblástico 3 y plakofilina 2, este último causal del 25 % de los casos. La mutación S358L que afecta al gen TMEM43 y causa la DAVD5, tiene penetrancia completa y es muy letal (OMIM 604400). Está en discusión si los casos con mutaciones dominantes en el receptor cardíaco de la rianodina sufren una DAVD o una TV polimórfica catecolaminérgica3,10,13.
También se ha descrito una forma autosómica recesiva de la DAVD asociada a queratodermia palmoplantar y pelo lanudo (enfermedad de Naxos; OMIM 601214), causada por mutaciones en homocigosis del gen de la placoglobina, cuyo producto es un componente de los desmosomas y las uniones celulares adherentes10,13.
La enfermedad de Uhl (OMIM 107970), defecto congénito poco frecuente con ausencia de miocardio ventricular derecho y pared del ventrículo derecho delgada como el papel, actualmente se considera una forma extrema de DAVD10.
La DAVD se manifiesta como una TV con morfología de bloqueo de rama izquierda del haz de His, que se origina en el ventrículo derecho, en adolescentes o adultos jóvenes aparentemente sanos. Las arritmias ventriculares suelen ser asintomáticas, pueden causar palpitaciones, síncope o MS. El reemplazo de músculo por tejido adiposo comienza por las áreas posterior e inferior del tracto de entrada del ventrículo derecho, adyacentes a la válvula tricúspide, al infundíbulo anterior y al vértice cardíaco, lo que forma el triángulo de la displasia. Funcionalmente causa anomalías de la contracción generales o regionales, alteraciones de la función sistólica/diastólica del ventrículo derecho, formación de aneurismas y dilatación e hipocinesia del ventrículo derecho. El tabique interventricular suele estar preservado, por lo cual las biopsias endomiocárdicas pueden no ser diagnósticas y la que se realiza en el ventrículo derecho tiene una sensibilidad baja, por lo segmentario de la infiltración grasa. También se han descrito zonas aisladas de sustitución grasa en pacientes ancianos, con el uso prolongado de corticoides, en la obesidad, en otras miocardiopatías y en la TV-TSVD idiopática13.
La interacción entre diferentes sustratos genéticos y los factores desencadenantes como el ejercicio extenuante o el entrenamiento enérgico, explican el espectro de formas de presentación clínica. Los principales factores que determinan una mala evolución son la disfunción grave del ventrículo derecho, la afección del izquierdo, el síncope, la edad temprana, el sexo masculino, los antecedentes de parada cardíaca, la TV rápida y mal tolerada, con diferentes morfologías, y la incidencia familiar de muertes súbitas juveniles. La limitación de la actividad física enérgica, pueden mejorar la evolución a largo plazo13.
4. Cardiopatías congénitas
En la evolución natural de las cardiopatías congénitas, así como tras la cirugía a corazón abierto, pueden presentarse trastornos del ritmo en un 40-50 % de los casos, lo cual se relaciona con los cambios de presión o volumen, a circuitos de reentrada creados por parches del tabique y líneas de sutura; que se expresan como taquicardias supraventriculares, aleteo y fibrilación auricular, o ambas. Los pacientes operados de tetralogía de Fallot tienen riesgo persistente de MS tardía con incidencia estimada de un 0,5 - 8,3 %13,23.
5. MS sin cardiopatía aparente
En las situaciones de MS sin cardiopatía aparente no se recogen evidencias de miocardio vulnerable por afectación orgánica o funcional y el factor desencadenante puede estar relacionado con el estrés físico o psíquico, seguramente por incapacidad de las técnicas actuales para detectar lesiones incipientes (miocarditis subclínica, desequilibrio del sistema nervioso autónomo no detectado, entre otros); pero debe suponerse que existe algún problema pues resulta difícil desencadenar fibrilación ventricular en un corazón sano3.
El síndrome de MS del lactante se ha asociado a posturas durante el sueño y recientemente se ha constatado hipofosfatemia como mecanismo desencadenante de los síntomas; luego de hiperfosfaturia que depleta el 50 % del fosfato en 24 horas y también, impide la liberación de oxígeno unido a la hemoglobina y causa asfixia. También se ha empleado el término SUID (sudden unexpected infant death) o MS inesperada en la infancia, para aquellos casos que mueren súbitamente antes de los 12 meses de vida y síndrome de MS infantil para el resto de los niños25,26.
La asociación con el sueño en decúbito prono y la exposición al humo del cigarro, se relacionan con polimorfismos de genes del sistema inmunológico y nervioso autónomo; y se estima que el 10 % están relacionados con canalopatías27.
EXPERIENCIA DE TRABAJO EN EL CARDIOCENTRO WILLIAM SOLER
En el Servicio de Genética Clínica se han evaluado tres familias con SQTL, una de ellas extensa con variabilidad intrafamiliar en la expresión clínica y en las detonantes identificadas para la MSC, pues algunos casos se presentaron durante la actividad física, otros durante el sueño y algunos fueron diagnosticados como epilépticos con cambios electroencefalográficos y compensados con tratamiento farmacológico.
Se realizó el estudio molecular con secuenciación de LQT1 y LQT3 a dos de sus miembros en edad pediátrica, uno sintomático y un hijo de padre afectado; pero no se detectaron mutaciones en estos genes. En esta familia hay casos con lesiones estáticas del sistema nervioso central por las MS, otros que han respondido a la terapia con betabloqueadores y otros con DAI (Figura 1).
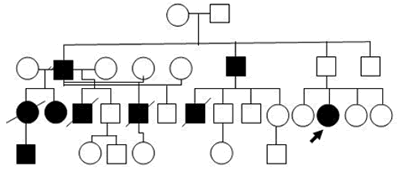
Figura 1. Árbol genealógico de familia con SQTL.
En cuanto a síndromes asociados a MS hubo una paciente menor de 1 año, con antecedentes familiares no relevantes, hija de padres no consanguíneos que tuvo desde el nacimiento sindactilia 3/4/5 (figura 2) desde la región proximal de los dedos hasta la distal en ambas manos (bilateral y simétrica), leve hipertelorismo ocular y comunicación interauricular tipo ostium secundum, sin repercusión hemodinámica. Tuvo un desarrollo motor normal sin clínica de inmunodeficiencia. Antes de cumplir el año de vida hizo MS durante el sueño que se recuperó y, a posteriori, se interpretó como un Síndrome de Timothy, posible nueva mutación.
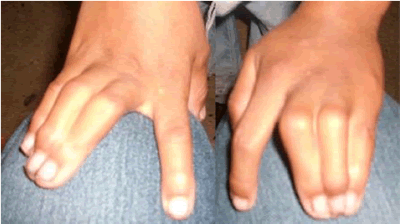
Figura 2. Sindactilia membranosa 3/4/5 similar a la de lactante fallecida con síndrome Timothy.
Los pacientes con miocardiopatías son evaluados por un protocolo que incluye estudios morfofuncionales cardiovasculares, pruebas metabólicas en orina, función hepática, amoníaco, prueba de tolerancia a glucosa combinada con ácido láctico, gasometría, y parcial de orina para descartar cetonuria. Se le realizan otros complementarios relacionados con las funciones inmunológicas, audición y visión, en dependencia de la causa sospechada, y se estudian 4 mutaciones relacionadas con miocardiopatías mitocondriales.
Se realiza la historia clínica genética que incluye árbol genealógico y examen físico, y se clasifica según el método clínico en aisladas no sindrómicas o asociadas a síndromes genéticos. A los familiares de aquellos casos con miocardiopatías aisladas no sindrómicas, sin antecedentes familiares, se les realiza ecocardiografía en busca de estigmas de miocardiopatías y también a aquellos con fenotipos sindrómicos como las rasopatías, que pueden tener hipertrofia segmentaria.
En este grupo de pacientes con MCPH y rasopatías, en especial el síndrome Costello que presenta ritmo caótico auricular como expresión electrofisiológica y de alto riesgo para MS, tuvimos la experiencia con un caso de 2 años de edad, diagnosticada desde el nacimiento con Síndrome Costello y MCPH, que requirió cirugía por la hipertrofia ventricular obstructiva con muy buena evolución postoperatoria, e hizo MS en el hogar unos meses después. Este caso fue interconsultado virtualmente con la Dra. Angela Lin, experta internacional en Síndrome Costello, quien corroboró el diagnóstico clínico y colaboró con el diagnóstico electrofisiológico, por lo raro de los trazados electrocardiográficos (Figura 3).
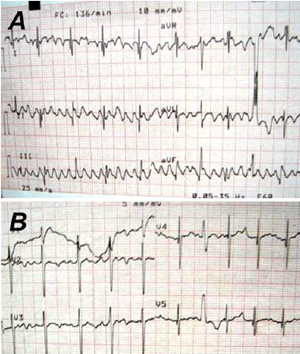
Figura 3. Electrocardiograma durante la lactancia de paciente con síndrome Costello, fallecida súbitamente.
Estas experiencias anecdóticas constituyen un acercamiento al trabajo clínico genético, que puede favorecer la prevención en otros miembros de la familia en riesgo, aun solamente al aplicar el método clínico.
Epílogo
La MSC se asocia a factores genéticos dados por los genes de susceptibilidad y predisposición, que constituyen condicionantes en interacción con factores ambientales desencadenantes para su expresión clínica. Esta asociación genoma-ambiente en la aparición de una enfermedad o fenotipo clínico es sinónimo de herencia multifactorial, aunque existan genes mayores o predisponentes con patrones de herencia bien definidos. Esas condicionantes genéticas no llegan a expresarse como enfermedad en muchos casos por interacciones epigenéticas endógenas o exógenas que dificultan el asesoramiento genético. La contribución de los estudios de correlación genotipo-fenotipo resulta esencial para esclarecer factores pronósticos, asociaciones terapéuticas así como acciones preventivas.
Nota del Editor
* El síndrome LEOPARD, también llamado lentiginosis múltiple, es una rara enfermedad de origen genético y transmisión hereditaria según un patrón autosómico dominante. El término LEOPARD es un acrónimo formado por las iniciales en inglés de sus principales manifestaciones (Lentigo, Anomalías en el Electroencefalograma, Hipertelorismo Ocular, Estenosis Pulmonar, Anomalías en los genitales, Retraso del crecimiento y Sordera, en inglés, Deafness)
Referencias bibliográficas
Bayés de Luna A, Elosua R. Muerte súbita. Rev Esp Cardiol. 2012;65(11):1039-52.
Ruskin J, McGovern B, Garan H. Sudden Cardiac Death. En: Jay H. Stein, Ed. Internal Medicine. 4ta Ed. Saint Louis: Mosby-Year Book Inc, 1994; p. 136-41.
Bayés de Luna A. Muerte súbita cardíaca. 1er Congreso Virtual de Cardiología [Internet]. Argentina; 1999 [consultado 2013 Oct 31]. Disponible en: http://www.fac.org.ar/cvirtual/cvirtesp/cientesp/chesp/chc5703c/cbayes.htm
Moya-i-Mitjans Á, Rivas-Gándara N, Sarrias-Mercè A, Pérez-Rodón J, Roca-Luque I. Síncope. Rev Esp Cardiol. 2012;65(8):755-65.
Ackerman MJ, Marcou CA, Tester DJ. Medicina personalizada: diagnóstico genético de cardiopatías/canalopatías hereditarias. Rev Esp Cardiol. 2013;66(4):298-307.
Real Academia Española. Diccionario de la lengua española [Artículo en Internet]. Madrid: Real Academia Española. [citado 2013 Oct 13]. Disponible en: http://lema.rae.es/drae/?val=susceptible
Singh VP, Rubinstein J, Arvanitis DA, Ren X, Gao X, Haghighi K, et al. Abnormal calcium cycling and cardiac arrhythmias associated with the human Ser96Ala genetic variant of histidine-rich calcium-binding protein. J Am Heart Assoc [Internet]. 2013 [citado 2014 Ene 11];2(5):e000460. Disponible en: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3835262/
Scholten A, Preisinger Ch, Corradini E, Bourgonje VJ, Hennrich MJ, , van Veen TAB, Swaminathan PD, Joiner ML, Vos MA, Anderson ME, Heck AJR. Phosphoproteomics study based on in vivo inhibition reveals sites of calmodulin-dependent protein kinase II regulation in the heart. J Am Heart Assoc [Internet]. 2013 [citado 2014 Ene 11];2(4):e000318. Disponible: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3828808/
Jiménez-Jáimez J, Tercedor-Sánchez L, Álvarez-López M, Martínez-Espín E, Sebastián Galdeano R, Almansa-Valencia I, et al. Estudio genético en el síndrome de QT largo en nuestro medio. Rev Esp Cardiol. 2011;64(1):71-4.
Hamosh A, Scott AF, Amberger JS, Bocchini CA, McKusick VA. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2005;33(Database issue):D514-7.
Jiménez-Jáimez J, Álvarez M, Algarra M, Macías Ruíz R, Peñas R, Valverde F, et al. Baja penetrancia clínica en sujetos portadores de mutación patogénica para las canalopatías cardiacas. Rev Esp Cardiol. 2013;66(4):275-81.
Jagu B, Charpentier F, Toumaniantz G. Identifying potential functional impact of mutations and polymorphisms: linking heart failure, increased risk of arrhythmias and sudden cardiac death. Front Physiol [Internet]. 2013 [citado 2014 Ene 11];4:254. Disponible en: http://journal.frontiersin.org/Journal/10.3389/fphys.2013.00254/full
Capulzini L, Brugada P, Brugada J, Brugada R. Arritmias y enfermedades del corazón derecho: de las bases genéticas a la clínica. Rev Esp Cardiol. 2010;63(8):963-83.
Benito B, Brugada J, Brugada R, Brugada P. Síndrome de Brugada. Rev Esp Cardiol. 2009;62(11):1297-315.
Brugada P. Epidemiología de la muerte súbita cardiaca. Rev Esp Cardiol. 2013;13(Supl. A):2-6.
Hsiao PY, Tien HC, Lo CP, Juang JM, Wang YH, Sung RJ. Gene mutations in cardiac arrhythmias: a review of recent evidence in ion channelopathies. Appl Clin Genet. 2013;6:1-13.
Almendral J, Castellanos E, Ortiz M. Taquicardias paroxísticas supraventriculares y síndromes de preexcitación. Rev Esp Cardiol. 2012;65(5):456-69.
de León Ojeda NE, Barreto García V, Ferraz Noda S. Propuesta de clasificación clínico-genética de los defectos cardiovasculares congénitos. Convención Internacional de Salud Pública. Cuba Salud 2012. [Artículo en Internet] La Habana, 3-7 de diciembre de 2012. [citado 2013 Oct 11] Disponible en: http://www.convencionsalud2012.sld.cu/index.php/convencionsalud/2012/paper/view/486/220
Laredo R, Monserrat L, Hermida-Prieto M, Fernández X, Rodríguez I, Cazón L, et al. Mutaciones en el gen de la cadena pesada de la betamiosina en pacientes con miocardiopatía hipertrófica. Rev Esp Cardiol. 2006;59(10):1008-18.
Pastore F, Parisi V, Romano R, Rengo G, Pagano G, Komici K, et al. Genetic test for dilated and hypertrophic cardiomyopathies: useful or less than useful for patients? Transl Med UniSa. 2013;5:14-7.
Cobo-Marcos M, Cuenca S, Gámez JM, Bornstein B, Ripoll T, García-Pavia P. Utilidad del análisis genético de la miocardiopatía hipertrófica en la práctica real. Rev Esp Cardiol. 2013;66(9):746-7.
Ackerman M, Priori S, Willems S, Berul C, Brugada R, Calkins H, et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies: this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Heart Rhythm. 2011;8:1308-39.
Gimeno JR, Oliva MJ, Lacunza J, Alberola AG, Sabater M, Martínez-Sánchez J, et al. Características de la muerte súbita en las cardiopatías hereditarias. Rev Esp Cardiol. 2010;63(3):268-76.
London B. Searching for sudden death SNPs in calcium handling genes. J Am Heart Assoc [Internet]. 2013 [citado 2013 Oct 23];2(5):e000541. Disponible en: http://jaha.ahajournals.org/content/2/5/e000541.long
Van Kempen TA, Deixler E, Crook MA. Hypophosphatemia as a key factor in sudden infant death syndrome (SIDS)? Ups J Med Sci. 2013;118(2):143-4.
Yoo SH, Kim AJ, Kang SM, Lee HY, Seo JS, Kwon TJ, et al. Sudden Infant Death Syndrome in Korea: A retrospective analysis of autopsy-diagnosed cases. J Korean Med Sci. 2013;28(3):438-42.
Wilders R. Cardiac ion channelopathies and the Sudden Infant Death Syndrome. ISRN Cardiol [Internet]. 2012 [citado 2013 Oct 23];2012:846171. Disponible en: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3529486/
Subir

















