



CorSalud 2014 Oct-Dic;6(4):341-345
CASO CLÍNICO
SÍNDROME DE MARFAN EN EDAD ADULTA: A PROPÓSITO DE UN CASO
MSc. Dr. Geovedy Martínez Garcíaa* y MSc. Dr. Gerardo Rodríguez Lemusb*
______________
Hospital Militar Central "Dr. Carlos J. Finlay". La Habana, Cuba.
Hospital Provincial "Agustino Neto". Guantánamo, Cuba.
* Colaboración Médica Cubana en la República Popular de Angola. Hospital do Prenda. Luanda, Angola.
Correspondencia: G Martínez García. Anita 936 e/ Gertrudis y Lagueruela, 10 de octubre. La Habana, Cuba.
Correo electrónico: geovedymtnez@infomed.sld.cu
Resumen
El síndrome de Marfan es una enfermedad del tejido conectivo, autosómica dominante, causada por defecto en el gen fibrilina-1, el cual desempeña un papel importante en la formación de los tejidos elásticos del cuerpo. Se diagnostica según datos clínicos, algunos de los cuales dependen del crecimiento. Nuestro objetivo es describir un caso atípico de síndrome de Marfan en un hombre de 44 años de edad, con antecedentes de salud, que acude al Servicio de Urgencias con disnea, dolores abdominales y edema intenso en miembros inferiores. En el examen físico se encontró soplo cardíaco, ingurgitación yugular y hepatomegalia congestiva. Se le realizó radiografía de tórax, ecocardiograma transtorácico y examen oftalmológico, y se llegó a la conclusión de estar en presencia de síndrome de Marfan, diagnóstico realizado en dependencia del cuadro clínico y de los criterios revisados de Ghent.
Palabras clave: Síndrome de Marfan, Insuficiencia aórtica, Alteraciones esqueléticas, Aneurisma aórtico, Subluxación del cristalino
Marfan syndrome in adulthood: a case report
Abstract
Marfan syndrome is an autosomal dominant connective tissue disorder, caused by a defect in the fibrillin-1 gene, which plays an important role in the formation of elastic tissues. It is diagnosed on clinical grounds, some of which depend on growth. Our purpose is to describe an atypical case of Marfan syndrome in a 44 year-old-male patient, with no history of health problems, who arrives at the Emergency Department with shortness of breath, abdominal pain and great pedal edema. On physical examination, heart murmur, elevated jugular venous pressure and congestive hepatomegaly were found. Chest radiograph, transthoracic echocardiogram, and ophthalmologic examination were performed, after which Marfan syndrome was diagnosed, according to the clinical picture and the revised Ghent criteria.
Key words: Marfan syndrome, Aortic regurgitation, Aortic aneurysm, skeletal disturbances, Lens subluxation
Introducción
El síndrome de Marfan es el trastorno hereditario multisistémico más común del tejido conectivo. Esta enfermedad, autosómica dominante, tiene una incidencia de 2-3 por 10.000 individuos, sin predilección de género, raza o etnia1. Se ha demostrado que la causa de esta enfermedad es un cambio en el gen FBN1, el cual codifica la proteína fibrilina-1, componente de la red de microfibrillas que sirve de armazón para el depósito de elastina y el ensamblaje de las fibras elásticas. En este gen se han descrito más de 500 mutaciones y casi todas son únicas para un individuo o familia afectada, lo que da origen a un defecto hereditario de la fibrilina-1, y ocasiona una formación de fibras elásticas anormales, con la consiguiente disfunción de los tejidos que la poseen2-10. Además, se ha postulado que la fibrilina normal inhibiría el crecimiento de los huesos largos y que las fibras elásticas a través de su tensión controlarían el crecimiento de estos, por lo tanto, al existir una alteración en estas estructuras se produciría el crecimiento óseo exagerado propio de la enfermedad.
Sus complicaciones afectan a ojos, pulmones y sistema musculoesquelético; pero la alta mortalidad de casos no tratados resulta casi exclusivamente de las complicaciones cardiovasculares, lo que incluye la disección aórtica y su ruptura5-6.
Los avances en el conocimiento de las causas del síndrome de Marfan, su diagnóstico temprano y la consecuente terapéutica médica o quirúrgica ha favorecido la mejoría marcada en el pronóstico de la población afectada, comparada con décadas anteriores. La identificación temprana de los pacientes asintomáticos es fundamental para reducir la frecuencia de los accidentes aórticos fatales. El presente trabajo tiene como objetivo presentar el caso de un paciente adulto con antecedentes de salud, que debutó con un cuadro clínico de insuficiencia cardíaca global, y que en el ingreso se le diagnosticó un síndrome de Marfan. Su importancia está dada por lo tardío del diagnóstico, que solo fue realizado en colaboración con la Brigada Cubana.
Caso clínico
Paciente masculino, de 44 años de edad, con antecedentes personales de salud, que acude a Urgencias del Hospital do Prenda, en Luanda, con falta de aire, la cual había comenzado hacía dos semanas, primero de esfuerzo, y luego lo obligaba a levantarse por las noches cuando estaba durmiendo, aliviándose al sentarse, con duración aproximada de una hora. También refería dolor abdominal en el hipocondrio derecho y aumento de volumen de miembros inferiores hasta la mitad de ambos muslos.
Datos positivos al examen físico
- Talla: 2,10 metros, brazos y dedos alargados. Aranodactilia. Signo muñeca-pulgar positivo. Laxitud articular, pie plano, y panículo adiposo disminuido (Figura 1).
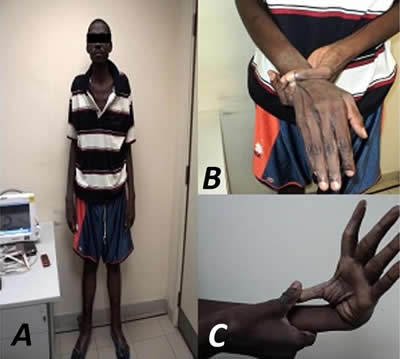
Figura 1. Características esqueléticas del paciente. A. Talla alta, brazos alargados, panículo adiposo disminuido. B y C. Aranodactilia, signo muñeca-pulgar positivo, laxitud articular.
-Pectus carinatum, con disminución de la expansibilidad torácica, polipnea superficial, frecuencia respiratoria de 34 por minuto y estertores crepitantes bibasales, más pronunciados en el hemitórax izquierdo.
- Pulso irregular y saltón. Choque de la punta visible, palpable y desplazado fuera de la línea media clavicular. Primer ruido disminuido en la punta, soplo sisto-diastólico en borde esternal izquierdo con predominio del componente diastólico (grado IV/VI), con irradiación al cuello. Ingurgitación yugular. Tensión Arterial: 100/70 mmHg.
- Abdomen blando y depresible, doloroso a la palpación en hipocondrio derecho, con hepatomegalia de 4 cm por debajo del reborde costal. Presencia de reflujo hepatoyugular.
- Edemas blandos, fríos y no dolorosos en miembros inferiores hasta la mitad de ambos muslos, con presencia de fácil godet.
Se decide ingresar al paciente con el diagnóstico de insuficiencia cardíaca global y se inicia tratamiento farmacológico, con el que se logra mejorar el cuadro clínico. Teniendo en cuenta las características fenotípicas de este paciente, así como los hallazgos auscultatorios sugerentes de enfermedad valvular aórtica, se decide realizar otros exámenes complementarios:
Ecocardiograma (Figura 2): Se observa aorta ascendente dilatada, de aspecto periforme, con diámetro de 77 mm, anillo aórtico no dilatado y cayado de la aorta normal (34 mm.). Regurgitación aórtica grave, que ocupa totalmente el tracto de salida del ventrículo izquierdo, con volumen regurgitante de 80 ml y orificio regurgitante efectivo de 0,95 cm². Ventrículo izquierdo dilatado, con hipocinesia global leve y función sistólica disminuida (fracción de eyección 40 %). Insuficiencias tricuspídea y mitral leves.
Examen oftalmológico: Lámpara de hendidura: en ambos ojos se observa córnea transparente, cámara anterior con profundidad aumentada, signo del sol naciente hora uno (subluxación del cristalino) e iridodonesis.
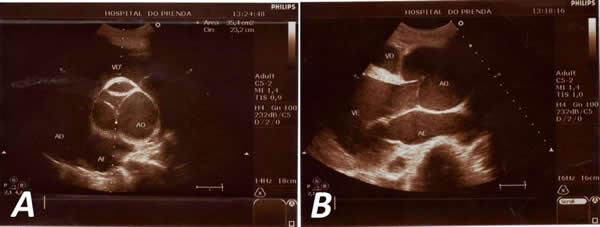
Figura 2. Ecocardiografía transtorácica. A. Eje corto a nivel de grandes vasos. Se observa la dilatación en raíz de la aorta con deficiente cierre de sus sigmoideas. B. Eje largo paraesternal. AD: aurícula derecha, AE: aurícula izquierda, AO: aorta, VD: ventrículo derecho, VE: ventrículo izquierdo.
Ante tales hallazgos se concluye que el paciente presenta un síndrome de Marfan. Se le plantea la posibilidad de tratamiento quirúrgico de la enfermedad valvular aórtica y acepta. Durante el ingreso en el hospital el paciente presenta parada cardiorrespiratoria y fallece.
Comentario
El síndrome de Marfan se produce por la alteración en las microfibrillas, causada generalmente por la mutación del gen FBN1, que codifica la fibrilina-1, y se localiza en el cromosoma 15q21; todo lo cual fue descrito inicialmente por Dietz et al.1, en 1991. La fibrilina-1, una glucoproteína extracelular esencial para la fibrinogénesis, es el principal componente de las microfibrillas de 10-12 nm, que junto con la elastina forman las fibras elásticas encontradas en los tejidos. Mutaciones del FBN1 incrementan la susceptibilidad de la fibrilina-1 para la proteólisis in vitro, con la fragmentación de las microfibrillas como consecuencia11,12.
El diagnóstico se basa fundamentalmente en los hallazgos clínicos. Existen tres formas de presentación relacionadas estrechamente con la edad, con cuadros clínicos y pronósticos bien definidos: neonatal, infantil y clásica. Esta última es la más frecuente y reconocida, y se presenta en niños, adolescentes y adultos. Conforme con los nuevos criterios diagnósticos para el síndrome de Marfan publicados en 2011, que surgen de la revisión de los clásicos criterios de Ghent, el fenotipo del paciente caracteriza el diagnóstico de este síndrome de otras enfermedades con fenotipos similares2.
Las alteraciones esqueléticas son las más frecuentes y precoces de detectar, y por ello son las que primero establecen la sospecha de la enfermedad. Son progresivas con la edad y se completan en la adolescencia. Destacan por su frecuencia la talla alta con mayor envergadura, pectum excavatum o carinatum, aracnodactilia, escoliosis, hipermotilidad articular y paladar ojival2,4,6-11. Las lesiones cardiovasculares determinan el pronóstico del síndrome de Marfan, ya que son las que ocasionan la mayor mortalidad, entre el 70-95 % de los casos, la más específica y frecuente ha sido la dilatación aórtica. Su incidencia depende de la edad, 40-80 % en niños y 80-100 %, en adultos13.
El compromiso ocular es frecuente (70 %) y progresivo. La lesión más específica para el diagnóstico es la subluxación del cristalino; sin embargo, es necesario también identificar vicios de refracción para preservar la máxima función visual14.
Al considerar los criterios diagnósticos antes descritos, en este paciente se encuentra:
Historia familiar y genética: se realizó examen físico a familiares de primer grado sin encontrar evidencia de la enfermedad.
Sistema esquelético:
Pectus carinatum: 2 puntos.
Pie plano: 1 punto.
Signo muñeca-pulgar: 3 puntos.
Sistema cardiovascular: dilatación de aorta ascendente con insuficiencia aórtica grave (presencia de criterio mayor).
Sistema ocular: subluxación del cristalino (presencia de criterio mayor).
Al excluir clínicamente los síndromes de Ehlers-Danlos y Loeys-Dietz, y teniendo en cuenta que no existían antecedentes familiares de la enfermedad, la presencia de dilatación de la raíz aórtica y de subluxación del cristalino permite el diagnóstico del síndrome de Marfan.
En la literatura revisada2-4,6 se mencionan todos los tipos clínicos de este síndrome, pero el diagnóstico de la enfermedad generalmente se realiza en la etapa infantil y en la adolescencia. Lo interesante del caso presentado es lo tardío del diagnóstico, en un paciente que tuvo varios ingresos por insuficiencia cardíaca y no contó con una evaluación completa y certera. En el momento actual se brinda asesoramiento genético a los otros miembros de la familia.
Referencias bibliográficas
Dietz H, Cutting G, Pyeritz RE, Maslen CL, Sakai LY, Corson GM, et al. Marfan syndrome caused by a recurrent do novo missense mutation in the fibrillin gene. Nature. 1991;352(6333):337-9.
Loeys BL, Dietz HC, Braverman AC, Callewaert BL, De Backer J, Devereux RB, et al. The revised Ghent nosology for the Marfan syndrome. J Med Genet. 2010;47(7):476-85.
von Kodolitsch Y, Robinson PN. Marfan syndrome: an update of genetics, medical and surgical management. Heart. 2007;93(6):755-60.
Lebreiro A, Martins E, Cruz C, Almeida J, Maciel MJ, Cardoso JC, et al. Marfan syndrome: clinical manifestations, pathophysiology and new outlook on drug therapy. Rev Port Cardiol. 2010;29(6):1021-36.
Sohn GH, Jang SY, Moon JR, Yang JH, Sung K, Ki CS, et al. The usefulness of multidetector computed tomographic angiography for the diagnosis of Marfan syndrome by Ghent criteria. Int J Cardiovasc Imaging. 2011;27(5):679-88.
Ammash NM, Sundt TM, Connolly HM. Marfan syndrome – diagnosis and management. Curr Probl Cardiol. 2008;33(1):7-39.
Jondeau G, Michel JB, Boileau C. The translational science of Marfan syndrome. Heart. 2011;97(15):1206-14.
Iams HD. Diagnosis and management of Marfan syndrome. Curr Sports Med Rep. 2010;9(2):93-8.
Chan YC, Ting CW, Ho P, Poon JT, Cheung GC, Cheng SW. Ten-year epidemiological review of in-hospital patients with Marfan syndrome. Ann Vasc Surg. 2008;22(5):608-12.
Ozdemir O, Olgunturk R, Kula S, Tunaoglu FS. Echocardiographic findings in children with Marfan syndrome. Cardiovasc J Afr. 2011;22(5):245-8.
Sponseller PD, Erkula G, Skolasky RL, Venuti KD, Dietz HC. Improving clinical recognition of Marfan syndrome. J Bone Joint Surg Am. 2010;92(9):1868-75.
Gao LG, Yao XP, Zhang L, Hui RT, Zhou XL. Molecular analysis for diagnosis of Marfan syndrome and Marfan-associated disorders. Chin Med J. 2011;124(6):930-4.
Pearson GD, Devereux R, Loeys B, Maslen C, Milewicz D, Pyeritz R, et al. Report of the National Heart, Lung, and Blood Institute and National Marfan Foundation Working Group on research in Marfan syndrome and related disorders. Circulation. 2008;118(7):785-91.
Nemet AY, Assia EI, Apple DJ, Barequet IS. Current concepts of ocular manifestations in Marfan syndrome. Surv Ophthalmol. 2006;51(6):561-75.
Recibido: 09 de junio de 2014
Aceptado: 03 de julio de 2014
Subir

















