CorSalud 2010;2(2)
CARDIOCENTRO "ERNESTO CHE GUEVARA"
SANTA CLARA, VILLA CLARA. CUBA
ARTÍCULO DE REVISIÓN
Síndrome de
Brugada: desde los genes hasta
Por:
Lic. Raimundo Carmona Puerta.
______________
Especialista de I Grado en Fisiología Normal y Patológica. Servicio de Electrofisiología Cardíaca Clínica y Estimulación. Cardiocentro Ernesto Che Guevara, Santa Clara, Villa Clara. Profesor Asistente. UCM-VC. e-mail: raimundo@cardiovc.sld.cu
Resumen
El creciente número de canalopatías iónicas descubiertas en el corazón con consecuencias fatales implica que los especialistas involucrados en el tratamiento de estos pacientes luchen por alcanzar un mayor conocimiento de la electrofisiología cardíaca básica. En el síndrome de Brugada ya se han descrito hasta seis de sus genotipos con afectaciones en los canales de sodio, calcio y potasio tipo Ito. En cualquiera de los casos se produce un electrocardiograma característico que aparece manifestado en derivaciones precordiales derechas, debido a la dispersión transmural de la repolarización más acentuada en la región del tracto de salida del ventrículo derecho. La enfermedad puede cursar asintomática o ser su primera manifestación la muerte súbita. El tratamiento más efectivo se considera el desfibrilador implantable, pero puede combinarse con quinidina para espaciar los choques y evitar tormentas eléctricas.
Abstract
The increasing number of ion channelopathies discovered in the heart, with fatal consequences, implies that the specialists involved in the management of these patients must strive to reach a better understanding of basic cardiac electrophysiology. In the Brugada syndrome, up to six genotypes have already been described with affectations in the sodium, calcium and potassium (Ito type) channels. In all cases there is a typical electrocardiogram which shows right precordial leads due to the transmural dispersion of repolarization, more pronounced in the region of the outflow tract of the right ventricle. The disease may be asymptomatic or have sudden death as its first manifestation. The implantable defibrillator is considered the most effective treatment, but it can be combined with quinidine to space the shocks and abort electrical storms.
Palabras clave: SIndrome de Brugada EstratificaciOn del riesgo Muerte súbita cardIaca Potencial de acciOn Heterogeneidad |
Key words: Brugada Syndrome risk stratification sudden cardiac death action potential heterogeneity |
Quizás no existan muchos ejemplos del vínculo ciencias básicas
– ciencias clínicas como el que ocurre en aquella subespecialidad de
Efectuemos entonces un profundo acercamiento a aquella alteración eléctrica que fuera una de las primeras en dejar de ser considerada primaria: el Síndrome de Brugada (SB).
Elementos básicos sobre fisiología molecular de la repolarización ventricular
Es difícil entrar en las intimidades de este síndrome sino se parte de la visión más avanzada de la repolarización ventricular.
Para garantizar la sucesión de latido a latido se necesita que un complejo de proteínas, conocidas como canales iónicos, funcionen perfectamente articuladas. Cada uno de estos canales trasmite una corriente mediante algún tipo de ion (sodio, potasio y calcio)1-3, y son importantes para comprender las bases iónicas del SB.
Canal de sodio (Nav)1,2
El nombre de la corriente de este canal es corriente de
sodio (INa). Posee una cinética de activación muy rápida,
y una cinética de inactivación y recuperación, rápida. Se ha encontrado en el corazón
de animales como: el gato, el perro, el hurón, el ratón, la rata y también, en humanos.
El canal por compuerta de voltaje Nav pertenece a la
superfamilia S4 de genes que codifican para estos canales. La subunidad α-Nav tiene cuatro dominios homólogos, cada uno de los cuales
atraviesa la membrana 6 veces. Los cuatro dominios se juntan para formar el poro central
selectivo al sodio (Na). Aunque existen homólogos de
α-Nav, Nav 1.5
codificada por el gen SCN5A es la subunidad α-Nav que más prominentemente se expresa
en el miocardio de mamíferos y es la causa de la fase 0 del potencial de acción
(PA) en miocitos atriales, ventriculares y células de Purkinje.
Sin embargo, existen algunos estudios que han demostrado la existencia de ARNm que codifica otras subunidades α-Nav, específicamente Nav 1.1, Nav 1.3 y Nav 1.4, las cuales típicamente
aparecen en el cerebro y el músculo esquelético, y se expresan además, en el
miocardio. Durante la meseta del PA en miocitos ventriculares humanos,
aproximadamente el 99 % de los canales Nav están en
estado inactivo no conductor, por tanto, existirá un 1 % de la población de
estos canales en estado conductor lo cual es conocido como componente tardío de
la corriente de Na (INa tardía).
Es por eso que hoy se acepta que
Canal de potasio (Kv)2
Pertenecen a la superfamilia S4 de canales por compuerta de voltaje. De las muchas subunidades α identificadas (Kv1-Kv9) solo unas pocas se expresan en el corazón. Es de nuestro interés según el tema expuesto, proporcionar algunos datos sobre la subfamilia Kv 4 que contiene a los canales transitorios de potasio. Estos poseen cinética rápida de activación, inactivación y recuperación, y se conocen como ItoF. En los grandes mamíferos parece que solo Kv 4.3 contribuye a ItoF. La corriente Ito determina la fase 1 del PA o repolarización rápida.
Canal de calcio (Cav)2,3
Pertenecen a la superfamilia S4 de genes que codifican para canales con compuerta de voltaje. Un canal Cav funcional es la resultante del ensamblaje multimérico de una α-Cav (α-1) y otras subunidades auxiliares (Cav ß y Cav α2γ), en algunos casos existen subunidades Cav γ. Este canal es un determinante significativo de la duración y morfología de la meseta del PA y, aunque muchas mutaciones extracardíacas aparecen en la literatura solo recientemente, se han encontrado en el miocardio provocando trastornos clínicos importantes. En el corazón se expresan los genes CACNA1C, CACNA1E, CACNA1G, CACNA1H, CACNB2, CACNA2D1 y CACNA2D2, que codifican respectivamente para las proteínas Cav 1.2, Cav 2.3, Cav 3.1, Cav 3.2, Cav ß-2, Cav α2γ-1 y α2γ-2.
Genética del Síndrome de Brugada
El modo de herencia es autosómico dominante. El primer gen relacionado al SB fue el SCN5A que codifica para α-Nav 1.5. Más de 100 mutaciones en este gen han sido descritas y solo una parte de estas se han estudiado en sistemas de expresión, lo que demuestra en todos los experimentos que la consecuencia final es la pérdida de la función. Los mecanismos mediante los que se produce esta pérdida de función son4-6.
- Alteración en la expresión del canal de Na.
- Cambio en las propiedades de dependencia en cuanto a tiempo y voltaje para la activación, inactivación o reactivación.
- Entrada del canal de Na en un estado intermedio de inactivación del cual se recupera lentamente.
- Inactivación acelerada del canal de Na.
Bezzina y colaboradores7 presentaron evidencias que apoyan la hipótesis de que los polimorfismos comunes en el promotor del gen SCN5A en asiáticos, modula la variabilidad en la conducción cardíaca y puede contribuir a la alta prevalencia del SB en la población de Asia.
Se ha informado un segundo locus en el cromosoma
3 cercano al SCN5A que está asociado a un trastorno progresivo de la
conducción, baja sensibilidad a la procainamida y un pronóstico
relativamente bueno, se considera que la mutación produce una reducción parcial
en
Existen dos genes más asociados al SB9. La mutación en ellos había sido predicha por la preparación de segmentos de miocardio ventricular derecho canino con bloqueadores del canal de calcio, que produce las manifestaciones arrítmicas y electrocardiográficas vistas en el SB y por observación, en seres humanos, de una forma adquirida de SB asociada a la ingestión de bloqueadores del canal de calcio. Dichos genes, son el CACNA1C y el CACNB2 que en los estudios genéticos y de expresión heteróloga revelaron pérdida de función en el canal de calcio tipo L. En los casos observados se combinó un fenotipo de SB con intervalos QT más cortos de lo normal9. También se han registrado mutaciones en los genes SCN1B y KCNE3 asociadas a esta alteración eléctrica (Tabla 1), que genera ganancia de función en la corriente ITO10,11.
Tabla 1 Mutaciones genéticas asociadas al Síndrome de Brugada.
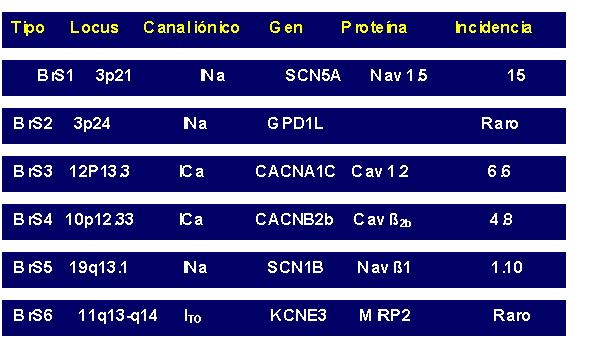
Tomado de Antzelevitch C, Nof E. Brugada syndrome: Recent advances and controversies. Curr Cardiol Rep. 2008;10:376-83.
Mediante métodos avanzados se ha predicho la existencia de proteínas relacionadas con la transmisión sináptica y el proceso fisiológico de la contracción muscular codificada por genes, cuya alteración podría producir un fenotipo de SB12. Quizás a partir de este punto se descubran nuevos genotipos de esta enfermedad.
Mecanismos de las arritmias en el Síndrome de Brugada
· El sustrato o circuito:
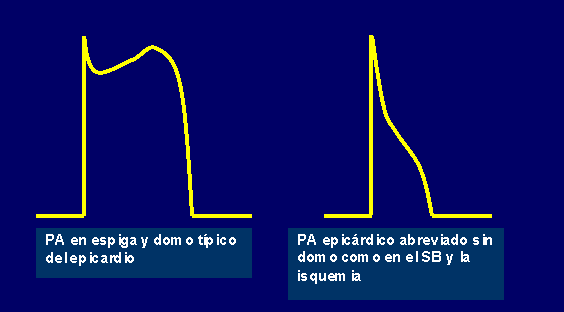
Está reconocido que el SB es una enfermedad del ventrículo derecho puramente eléctrica, por tanto aquí hablaremos de un circuito funcional. Dicho circuito estaría facilitado por la significativa expresión de la corriente ITO en el epicardio del tracto de salida del ventrículo derecho (TSVD)13 que existe normalmente. Cuando diversas circunstancias producen acentuación de ITO, la duración del PA (DPA) se acorta y se produce un fenómeno conocido como decapitación o pérdida del domo del PA13-16. Esto ha sido reconocido como repolarización todo o nada (Fig. 1).
|
Figura 1 Comparación entre dos potenciales de acción epicárdicos en condiciones normales (a la izquierda) y como se manifiesta en el SB (a la derecha). |
El sustrato se establece sobre la base de una exacerbación de la heterogeneidad intrínseca que existe en el miocardio normal13, ya que las alteraciones descritas en el epicardio no ocurren en el endocardio, por tanto se crea una ventana vulnerable transmural. Esta misma dispersión transmural de la repolarización (DTR) es la base de los cambios electrocardiográficos observados en estos pacientes14. También existen autores que intentan asociar elementos estructurales como favorecedores de las arritmias en el SB. Se ha encontrado conducción lenta en el TSVD, con presencia de potenciales ventriculares tardíos y electrogramas fragmentados de baja amplitud y tardíos, que contribuyen a la inducción de FV mediante la estimulación eléctrica programada; la base de estos hallazgos en parte podría deberse a alteraciones patomorfológicas como infiltración del tejido graso, fibrosis intersticial, infiltración linfocítica y/o desorganización miocítica, según el análisis de biopsias endomiocárdicas informadas en un reciente estudio17.También se han demostrado alteraciones estructurales atriales que parecen asociarse a los casos de SB de más alto riesgo18. Aunque la mayor parte de los investigadores considera que el ECG tipo 1 es producido por la característica heterogeneidad de la repolarización observada en estos pacientes, Postema y colaboradores consideran que los trastornos de la despolarización del ventrículo derecho expresados como un QRS más ancho en precordiales derechas y demoras tardías de la conducción en dicha cámara, son los cambios primarios y las anomalías de la repolarización que deben verse como alteraciones secundarias al trastorno de la despolarización19.
- El disparador:
No solo existe un aumento en
|
Figura 2 Esquema que muestra, de forma sencilla, el fenómeno de la reentrada de fase 2. |
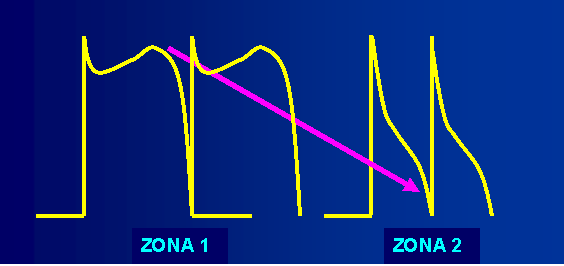
Representación de dos zonas cercanas en el epicardio del TSVD con sucesos eléctricos que ocurren casi al unísono. Obsérvese que se crea un vector de corriente desde la zona 1 en el momento que ocurre el domo del PA (Fase 2) que captura al tejido de la zona 2, unos milisegundos más tarde el domo del segundo PA, inducido en la zona 2, crea otro vector de corriente dirigido a la zona 1 que produce la captura de ese tejido que ya está completamente recuperado, y se genera un segundo PA en la zona 1.
La reentrada de fase 2 recibe ese nombre porque es inducida por el domo del PA que ocurre en fase 2. Este tipo de reentrada puede producir extrasístoles y salvas cortas de taquicardia ventricular, que pueden penetrar en la ventana vulnerable transmural para dar inicio a la típica taquicardia ventricular polimórfica (TVP) que ocurre en este síndrome. Los estudios en preparaciones de animales habían predicho que las extrasístoles reentrantes de fase 2 debían poseer tiempos de acoplamiento muy cortos13-15; sin embargo, en el trabajo de Thomsen y colaboradores con humanos, se observó que el rango de los tiempos de acoplamiento osciló desde 300-505 ms, incluso la mitad de los sujetos mostraron acoplamientos de 400 ms o mayores21. Antzelevitch ha sugerido que probablemente el inicio de la reentrada de fase 2 sea oculta en la onda T del latido sinusal precedente y considera que solo el segundo latido es claramente discernible en el ECG y produce, el aparentemente largo intervalo de acoplamiento observado en humanos22.
Rol del sistema nervioso autónomo (SNA) en la arritmogénesis del SB
La taquicardia ventricular, la muerte súbita o ambas, en el SB usualmente, ocurre en reposo, muchas veces en la noche mientras se duerme, lo que sugiere un patrón circadiano asociado al balance simpatico-vagal22. Los neurotransmisores autonómicos como la acetilcolina, facilitan la pérdida del domo del PA por supresión de la corriente de Ca y aumento de la corriente de potasio6,13. Los agonistas ß-adrenérgicos restauran el domo por aumento de la corriente de Ca6,13, es por eso que durante un protocolo de estimulación programada para inducir arritmias en un supuesto paciente con SB jamás estará indicado el isoproterenol, a diferencia de cuando se estudia cualquier otra arritmia.
Otros moduladores y perpetuadores en la arritmogénesis del SB (Fig 3).
|
Figura 3 Moduladores y perpetuadores en la arritmogénesis del SB. |
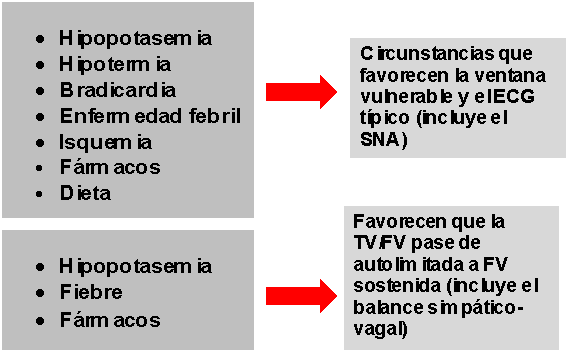
- Hipotermia23,24: acentúa marcadamente la melladura de la fase 1 del PA, lo que lleva a la pérdida del domo en algunas regiones del epicardio. Lo anterior crea un aumento del gradiente de voltaje transmural responsable de la aparición o exacerbación de la onda J o elevación del segmento ST. Se favorece con esto la aparición de extrasístoles y colgajos breves de taquicardia ventricular.
- Hipopotasemia25: se conoce que la mayor prevalencia del síndrome de la muerte súbita inexplicable nocturna (ahora demostrado ser el SB), es en el noreste de Tailandia cuando se compara con Bangkok, que se encuentra en la parte central del país, y ocurre porque la deficiencia de potasio es endémica de la región del noreste, con niveles de potasio significativamente menores entre sus habitantes respecto a los de la capital.
- Fiebre: los sistemas de expresión heteróloga son usados frecuentemente en los estudios de mutaciones de canales iónicos. Los
investigadores señalan que a la temperatura de habitación en la que suelen
efectuarse estos experimentos (22o C), las alteraciones de función
en el canal de sodio se manifiestan en menor medida que a temperatura fisiológica13. Debido a esto la pérdida
de función del canal mutante es más evidente a temperaturas por encima del
rango fisiológico4. Desde hace algunos años existe la demostración clínica
de pacientes con SB que han desarrollado episodios de TVP durante la fiebre26.
También se han mencionado a los baños calientes como factor precipitante.
Recientemente se registró una paciente de 18 años con mastitis, fiebre y
síncope, su ECG mostró un patrón tipo 1 de SB que desapareció al terminar la
fiebre27. También se informa
el descubrimiento de la enfermedad en un hombre de 37 años con fiebre, causada
por una shigellosis gastrointestinal tras la
ingestión de comida china. Las alteraciones electrocardiográficas compatibles
con SB desaparecieron junto con la fiebre28. Samani y colaboradores develaron una nueva mutación que predispone a
- Isquemia
miocárdica: se considera al SB un modelo estable (no isquémico) de los
cambios que ocurren durante la lesión isquémica aguda13. El infarto miocárdico o la isquemia aguda secundaria
a vasoespasmo en la región del TSVD imita una
elevación del segmento ST similar al observado en el SB22. Particularmente las lesiones o el vasoespasmo de la pequeña arteria del cono pueden desenmascarar
este tipo de ECG31. Esto se debe a depresión
de la corriente de calcio tipo L y a la activación de la corriente de potasio
dependiente de ATP (IKATP), lo anterior predispone a pacientes con
SB a un mayor riesgo de muerte cardíaca súbita relacionado con la isquemia22. Recientemente Hu y colaboradores32 estudiaron a 19 pacientes con infarto agudo de miocardio, que desarrollaron complicaciones
arrítmicas ventriculares, 18 de estos presentaron 1-2 episodios de FV mientras
que solo uno presentó una tormenta eléctrica (6 episodios en las primeras 12
horas)33.
Este paciente fue el único en el que se demostró una mutación en el gen SCN5A
producida por una sustitución de un aminoácido por otro (G400A), además en el
mismo alelo se encontró el polimorfismo H558R, existió una reducción en el valor
máximo de INa de 88,4 % respecto a los controles a
-35 mV. Se considera esta la primera mutación en el
canal de sodio asociado a tormenta eléctrica durante la isquemia aguda. Cordeiro y colaboradores encontraron que la disponibilidad
reducida de
- Fármacos: queda pendiente definir cuando estamos en presencia de un SB solapado, desenmascarado por fármacos, o un signo de Brugada. Se conocen múltiples medicamentos y tóxicos capaces de producir formas adquiridas de SB o exponer fenotipos ocultos. No existe idea en la comunidad científica de cuándo terminaremos de completar una lista con dichas sustancias. Una versión reciente del mencionado listado fue elaborada por Antzelevitch y Viskin15, pero constantemente aparecen informes de nuevas sustancias. Sin embargo, no encontraremos allí un fármaco muy utilizado en nuestro medio y que recientemente fue asociado a MCS y un ECG tipo 1 de SB35: el propofol, uno de los anestésicos más empleados en la medicina moderna. Vernooy y colaboradores36 revisaron los datos clínicos de 67 pacientes ingresados en la unidad de cuidados intensivos neuroquirúrgicos del University Medical Center Utrecht, en Holanda entre los años 1996-99 y que recibieron infusión prolongada de propofol, entre estos se identificaron 7 con el síndrome por infusión de propofol. Seis de ellos, y ninguno de los restantes, presentaron un ECG tipo 1. Todos los casos con dicho ECG desarrollaron arritmias ventriculares que degeneraron en FV irrecuperable, la dosis usada en el momento que se registró en el ECG fue 7,9 ± 1,2 mg/kg/h. Los estudios en cuñas perfundidas indican que la elevación del ST inducido por propofol no es el resultado de un efecto directo del medicamento durante la exposición a corto plazo de altas concentraciones de este. Los autores consideran que un metabolito, la presencia de anomalías electrolíticas, acidosis, fiebre, o la combinación con otros fármacos, contribuyen al desarrollo del patrón electrocardiográfico y a las arritmias. También destacan que es clara la relación de la muerte súbita arritmogénica con el anestésico, pues describen su más reciente experiencia con un paciente en el que tras identificar el ECG tipo 1 detuvieron la infusión y todos los cambios desaparecieron, además de no ocurrir arritmias. Vernooy y colaboradores concluyeron que si en el curso de una infusión de propofol aparece un ECG similar al del SB debe interpretarse como inestabilidad eléctrica y predice una muerte cardíaca inminente.
Otro anestésico muy usado en nuestro medio de forma local o como antiarrítmico, y que tampoco aparece en ningún listado de fármacos inductores de un patrón electrocardiográfico similar al Brugada, es la lidocaína. Un estudio reciente de Barajas-Martínez y colaboradores37 informaron el caso de un paciente de raza negra sin historia de enfermedad cardíaca, que llegó a un cuerpo de guardia tras una convulsión. Presentó un ECG inicial normal hasta que irrumpió un colgajo de taquicardia ventricular monomórfica con deterioro hemodinámico, que fue tratado con 70 mg de lidocaína en bolo, seguido de infusión continua a 1mg/min, combinado con un choque eléctrico. Tras la administración del medicamento se manifestó una elevación del segmento ST > 2mm de V1-V3 con enzimas normales. El paciente fue diagnosticado como SB por la presencia de una arritmia maligna y la aparición de un ECG tipo 1 con el uso de un bloqueador de canales de sodio. Un análisis ulterior de posibles mutaciones en el gen SCN5A demostró el cambio de un aminoácido por otro en dos posiciones diferentes (valina 232 isoleucina y leucina 1308 fenilananina), capaces de reducir la corriente de sodio un 73 ± 0,09 % cuando se asocian a la administración de lidocaína respecto al uso de lidocaína en los controles (reducción del 18,2 ± 0,1 %). Los autores consideran que esta doble mutación hace que la lidocaína se comporte como un fármaco de las clases IA o IC indicados para desenmascarar fenotipos del SB.
Como novedades se informó que el litio (indicado en trastornos bipolares) puede desenmascarar un SB en concentraciones subterapéuticas38, y también, un reciente antidepresivo tricíclico es capaz de producir un ECG tipo Brugada39. Dicho fármaco es la dosulepina y se considera capaz de alterar la sensibilidad de la prueba de ajmalina, pues el caso registrado por Meert y colaboradores fue positivo cuando se aplicó 4 días después de suprimir el fármaco, pero cuando se repitió la prueba 11 días después del retiro, fue negativa.
SB y fibrilación auricular (FA)
Además de las ya conocidas TVP y FV que producen
los efectos letales del SB podemos encontrar FA asociada a esta enfermedad6. Con frecuencia no solo aparece
más FV durante el sueño en estos pacientes, sino también más FA, el mecanismo básico
parece ser la disminución del tono simpático más que un aumento del tono vagal, este efecto conlleva a una menor disponibilidad de
calcio aportado por ICaL, lo
que favorece la pérdida del domo del potencial de acción40.
Recordemos que la corriente ICaL está
poderosamente regulada por el sistema nervioso simpático.
Diagnóstico del Síndrome de Brugada
Viene determinado por lo establecido en el registro
de
Tabla 2 Fármacos aprobados para desenmascarar un ECG diagnóstico:
Fármaco |
Dosis |
Vía de administración |
Ajmalina |
1 mg/kg a pasar en 5 min |
Intravenosa |
Flecainida |
2 mg/kg a pasar en 10 min 400 mg |
Intravenosa Oral |
Procainamida |
10 mg/kg a pasar en 10 min |
Intravenosa |
Pilsicainida |
1 mg/kg a pasar en 10 min |
Intravenosa |
- FV documentada
- TVP
- Historia familiar de muerte cardíaca súbita en personas < 45 años
- Electrocardiograma tipo 1 en miembros de la familia
- Taquicardia ventricular inducida por estimulación eléctrica programada
- Síncope
- Respiración agónica nocturna
El síncope debe interpretarse con suma cautela antes de darle un valor definitivo en el escenario del SB. Kostopoulou y colaboradores43 encontraron en su estudio una prevalencia de la prueba de la mesa basculante positiva, sensibilizada con clomipramina, del 75 % (15 de 20 casos), lo que sugiere que estos individuos son susceptibles de padecer síncope vasovagal y estar sujetos a la confusión sobre la agresividad de su enfermedad, ya que en estos casos el síncope no es de origen arrítmico, lo que puede llevar a una decisión errónea acerca del pronóstico de su enfermedad. Los autores no encontraron correlación entre el desarrollo de las arritmias ventriculares sostenidas y el prueba de la mesa basculante positiva.
El uso de estos fármacos se basa en que el ECG
tipo 1 de SB es frecuentemente oculto y se manifiesta intermitentemente de
forma espontánea o bajo condiciones especiales que pueden ser simuladas por estos
fármacos. Otras veces no está presente el ECG tipo 1 pero existe sospecha de la
enfermedad en presencia de las variantes tipo 2 y
Tabla 3 Tipos de ECG descritos en pacientes con SB.
Tipo 1 |
Tipo 2 |
Tipo 3 |
|
Punto J |
≥2 mm |
≥2 mm |
≥2 mm |
Onda T |
Negativa |
Positiva o bifásica |
Positiva |
Configuración del ST-T |
Convexa |
En silla de montar |
En silla de montar |
Porción terminal del segmento ST |
Descenso gradual |
Elevado ≥ 1 |
Elevado < 1 |
Los criterios de un ECG tipo 1 para ser válidos en el diagnóstico de SB, deben estar presentes en más de una derivación precordial derecha desde V1-V3. No obstante, debemos tener presente que aunque se menciona poco, existen variantes actualmente consideradas similares al Síndrome de Brugada en los cuales existen todas las características de un ECG tipo 1 salvo que se manifiestan en derivaciones de cara inferior o lateral izquierda, incluso acompañando a mutaciones del gen SCN5A6. Sarkozy y colaboradores informaron, por primera vez, 3 casos de SB en quienes se manifestó el ECG tipo 1 tras la administración de bloqueadores del canal de sodio en derivaciones inferiores44-46.
Miyamoto y colaboradores47 demostraron que existen pacientes con diagnóstico confirmado de SB mediante pruebas genéticas, que no exhiben un ECG tipo 1 en sus registros basales cuando se aplican los electrodos en posición convencional, y que al desplazarlos hasta el segundo espacio intercostal, muestran el ECG diagnóstico con un pronóstico similar a aquellos pacientes con el típico ECG del SB. Estos autores recomendaron efectuar este procedimiento antes que aplicar antiarrítmicos con el fin de desenmascarar un ECG tipo 1, pues sería lo más apropiado debido a que es una técnica sencilla y exenta de riesgo. El uso de fármacos quedaría limitado para aquellos pacientes en los que esta conducta no fuera útil. Recientemente se propuso obtener derivaciones precordiales bipolares mediante una combinación de V2 (polo +) y V4 o V5 (polo -), ya que estas derivaciones [V (2-4) y V (2-5)] son más sensibles que V2 (en el cuarto espacio intercostal) para la detección de pacientes con SB48
No puede olvidarse que, además de todo lo expresado en el proceso de diagnóstico del SB, tenemos que descartar otras condiciones capaces de producir un ECG similar al del SB.
Estimación del riesgo
El riesgo en el caso de esta enfermedad conduce a la decisión de insertar un DAI o realizar seguimientos periódicos por consulta. Los temas de discusión están centrados en el SB asintomático y en el rol del estudio electrofisiológico (EEF), como estratificador de riesgo en estos pacientes. Por otra parte, se mantiene una amplia aceptación respecto a la implantación de un DAI en casos de SB que experimentan arritmias potencialmente letales, MCS recuperada o respiración agónica nocturna. Es por eso que centraremos la discusión de este tópico en los temas controversiales.
Experiencia de los doctores Brugada comparada con la de otros autores:
Brugada y colaboradores después de analizar 547 individuos diagnosticados con SB sin paro cardíaco previo concluyeron6:
- Un ECG tipo 1 posee un riesgo 7,7 veces mayor de desarrollar arritmias, en comparación con aquellos sujetos en los que el diagnóstico de la enfermedad solo pudo efectuarse al usar un bloqueador de canales de sodio.
- Ser varón implica un riesgo 5,5 veces mayor de la ocurrencia de muerte súbita respecto a mujeres.
- La estimulación eléctrica programada que induce a arritmias ventriculares sostenidas constituye el más poderoso marcador de riesgo, pues tiene 8 veces más riesgo de padecer muerte súbita que aquellos pacientes en los que no sean inducibles.
- Las formas familiares de SB no están asociadas a un peor pronóstico respecto a los casos esporádicos.
Se ha sugerido que el protocolo de estimulación
que debe aplicarse en estos pacientes debe contener hasta un máximo de 3 extraestímulos
a longitudes de ciclo ≥ 200 ms, estimulando desde el ápex del ventrículo
derecho. En el caso que no se induzcan arritmias debe ser repetido desde el
TSVD25. Sin embargo, la inducción en la práctica no es tan alta, lo
que puede generar conclusiones fatales; es contradictorio que cuando se
estudian porciones de miocardio ventricular perfundidas por vía coronaria, sea fácil
la inducción de arritmias ventriculares, la mayoría de las veces con un solo extraestímulo (S2), lo que crea una aparente discrepancia
entre resultados básicos y clínicos. Precisamente conocer la fisiología básica
que gira alrededor de esta enfermedad nos permite derrumbar tal aparente
discrepancia. En electrofisiología cardíaca clínica usualmente estimulamos el
endocardio, en cambio las porciones de miocardio son estimuladas desde el
epicardio, lo que produce una inversión en la activación miocárdica. Dicha inversión
significa que la región con períodos refractarios más cortos en los ventrículos
(epicardio), sea la primera en despolarizarse, de forma tal que cuando el
impulso eléctrico llegue a la región M y a los miocitos subendocárdicos ya el epicardio estará completamente recuperado. Esto produce un incremento en
la ventana vulnerable (aumento en
Un análisis de regresión logística aplicada a la serie de los Brugada permitió definir 8 categorías de riesgo, de los cuales los pacientes asintomáticos con ECG basal normal y FV no inducible por EEF, constituyen la población de más bajo riesgo, y los pacientes con síncope, ECG tipo 1 espontáneo e inducción en el EEF, poseen el peor pronóstico16. Es importante destacar que existe una clara disminución en el registro de la tasa para un primer suceso de FV en la serie de los Brugada, en 1998 informaron que el 27 % de pacientes previamente asintomáticos experimentaron un primer episodio de FV o MCS; sin embargo, ya en el año 2005 informaron que esto solo sucedió en el 6 % de los casos en un seguimiento medio de 42 ± 42 meses, con una tasa del 1,7 %10. Ellos mismos consideran que quizás los primeros casos que formaron parte de su registro fueron variantes muy agresivas del síndrome, lo que sobreestima la interpretación del riesgo adjudicada a los casos asintomáticos.
En el reciente metaanálisis de Paul y colaboradores con más de 700 pacientes, incluidos en más de 15 estudios que además, consideraba más de 400 casos de la serie Brugada para establecer comparaciones, no se pudo atribuir características de los pacientes fáciles de identificar que explicaran la mayor inducción observada en la serie Brugada, ya que el porcentaje de hombres y de casos con elevación espontánea del ST tipo convexa fue similar en ambos grupos. Tampoco podríamos implicar a la agresividad del protocolo de estimulación empleado, ya que los datos demuestran que los doctores Brugada siempre estimularon desde el ápex del ventrículo derecho, mientras que en los otros estudios se utilizó también la estimulación desde el TSVD, lo cual debería duplicar la posibilidad de inducir FV.
Existe una coincidencia entre todos los estudios en cuanto al valor predictivo negativo (considerado muy útil) de la estimulación programada, con un rango de ocurrencia de FV espontánea de 1 % en la serie Brugada y 2 % entre los pacientes de los 15 estudios incluidos en el metaanálisis de Paul y colaboradores en los casos no inducibles50. También está claro que el conflicto se encuentra en el valor predictivo positivo51. Priori y Eckard encontraron una tasa de sucesos anuales de 0,8 % - 1 % en pacientes inducibles, que sugerían que la estratificación del riesgo basada en la inducibilidad por EEF era pobre52,53. La mayoría de los estudios que analizaron la importancia del EEF en la predicción futura de riesgo en casos asintomáticos concuerdan en la incapacidad de esta prueba con tal fin. La más grande serie del mundo publicada hasta el momento, el registro FINGER54 que involucró 4 países de Europa (Francia, Italia, Holanda y Alemania) con 1029 pacientes consecutivos, seguidos por un promedio de 34,9 meses (14-54,4), mostró una tasa de complicaciones cardíacas por año de 7,7 % en pacientes con MCS recuperada, 1,9 % en casos con síncope y 0,5 % en asintomáticos. También demostró, como otros trabajos, que la inducción de taquiarritmia ventricular no es predictora de complicaciones eléctricas cardíacas, al igual que una historia familiar de MCS. En un articulo reciente de Viskin y colaboradores, se reconoce que es compleja la interpretación de un EEF positivo en este tipo de enfermedad, si se parte de la premisa de que es posible inducir FV con 2-3 extraestímulos ventriculares en al menos 6 % de los individuos sanos, incluso esta cifra pudiera llegar hasta 41 % ya que en este valor se incluyen sujetos que hicieron TVP no sostenida y en los cuales no se continuó el protocolo de estimulación, para así evitar el choque con corriente directa55. Esto quiere decir que existen muchos casos falsos positivos entre los Brugada asintomáticos inducibles por EEF y a los que se les implantará un DAI que jamás funcionará.
Kamakura y colaboradores
examinaron el pronóstico de individuos con comprobación genética de SB y ECG
ón del ST ≥
Otros factores de riesgo que predicen eventos arrítmicos en el SB10:
- Anchura de la onda Sa y magnitud de elevación del segmento ST
- Potenciales tardíos
- Prolongación en la duración del QRS (> 102 ms) en V2 o un intervalo rJ en V2 ≥ igual a 90 ms
- Neuropatía autonómica
- Intervalo Tmáxima-Tfinal y su dispersión
- Intervalo QTc > 460 ms en V2
- Alternancia de la onda T, especialmente tras la exposición a bloqueadores del canal de sodio b
- Presencia del signo de AvR (onda R prominente en la derivación AvR)
- Fibrilación auricular espontánea41
- Presencia de una repolarización precoz en la cara inferior46,56 y lateral46
- Retardo en el comienzo de la contracción entre el ventrículo derecho y el izquierdo
- ECG tipo 1 espontáneo en V257
- QRS fragmentadoc
- Síndrome del nodo SA enfermod
a La onda S en DII y DIII es significativamente más ancha en pacientes con una prueba positiva al urtilizar bloqueadores del canal de sodio que en aquellos con un resultado negativo58. La ausencia de onda S en derivaciones laterales izquierdas con elevación del ST en precordiales derechas ayuda a excluir un verdadero trastorno de la conducción intraventricular derecha10.
b En el escenario del SB se debe a dos mecanismos distintos59:
1. Pérdida del domo en latidos alternantes
2. Reentrada oculta de fase 2 en latidos alternantes
El grado de alternancia parece predecir la aparición de arritmias letales.
c El valor predictivo de MCS de este parámetro puede incrementarse si se combina con otra variable que indique repolarización anormal como la microalternancia de la onda T60.
d Al momento de confección de este artículo constituye el más reciente predictor de riesgo para el desarrollo de FV, pero tiene la limitante que solo es útil en esta subpoblación de sujetos y cobra fuerza si existe un ECG tipo 1 asociado. A su vez la prevalencia de SB es notablemente superior en esta bradiarritmia con respecto a la población general, lo que nos obliga a buscar con mayor fuerza el SB dentro de estos pacientes61.
Además de Kamakura y colaboradores, Sarkozy y colaboradores, encontraron un patrón de repolarización precoz en derivaciones inferiores y laterales con relativa frecuencia, que se asoció a un fenotipo más severo de la enfermedad, lo que ratifica que este puede usarse como variable pronóstica46. Otra interesante variable pudiera ser la prueba del estómago lleno (consiste en un almuerzo fuerte obviando el desayuno durante un intervalo no mayor de 20 minutos y realizar registros electrocardiográficos en los primeros 30 minutos posprandiales, es positiva si se registra un ECG tipo 1. Se aplica en casos sin ECG tipo 1 al momento de la prueba) que cuando es positiva se asocia con arritmias potencialmente letales hasta en el 87 % de los casos y en 17 %, cuando la prueba es negativa según un trabajo de Ikeda y colaboradores62.
Terapéutica del SB
El DAI constituye hasta el momento la única
terapia con mayor efectividad en el SB. Su indicación se plantea acorde a lo
sugerido en el informe de
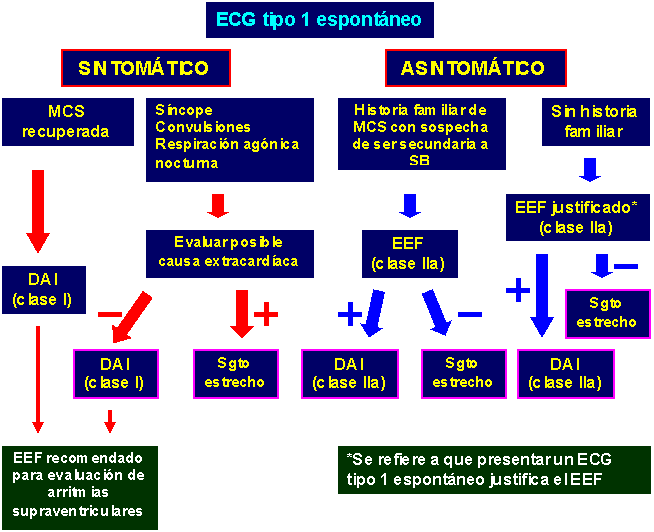
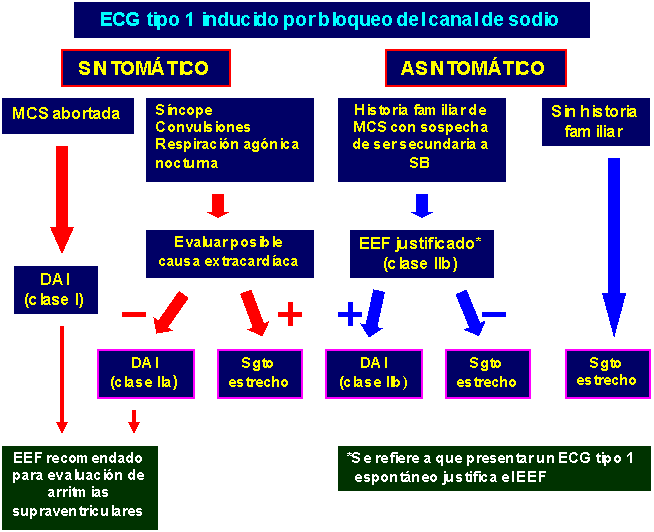
La efectividad del DAI para revertir
No obstante, aunque surgió esta nueva propuesta de programación es válido considerar terapias alternativas y coadyuvantes según la situación a enfrentar. Principalmente, se han estado buscando opciones farmacológicas que puedan combinarse con el DAI para prevenir choques frecuentes y mejorar la calidad de vida de los pacientes, además de que constituyan un arma para detener tormentas eléctricas. La posible utilización del marcapasos para prevenir frecuencias lentas ha sido mencionada, pero hasta el momento no existen evidencias que justifiquen su uso6.
Con respecto a la criocirugía o ablación focal, como técnicas que destruyen focos arritmogénicos causales de extrasístoles disparadoras de TV/FV, no existen datos hasta el momento de peso suficiente como para recomendarlos como alternativas de uso regular63.
Dentro de las opciones farmacológicas existe una
sin rival, la quinidina. Se ha sugerido como un agente efectivo para suprimir
FV en el SB, al inhibir la corriente ITO, lo cual reduce el grado de
heterogeneidad de la repolarización en la región del TSVD65. La quinidina
es el único agente disponible en el mercado con significativas propiedades
bloqueantes de ITO66. Ha resultado eficaz en la prevención
de
Otras opciones que aparecen en la literatura:
- ISOPROTERENOL: solo o en combinación con quinidina ha probado ser efectivo en la normalización del ST en casos con SB y en el control de tormentas eléctricas, especialmente en niños 63. Este fármaco reduce el tono vagal e incrementa la frecuencia cardíaca, lo que contribuye a la reducción de ITO10.
- CILOSTAZOL: es un inhibidor de la fosfodiesterasa III con resultados contradictorios, en un
estudio previno
- 4-AMINOPIRIDINA: teóricamente una sustancia ideal para bloquear de manera exclusiva la corriente ITO. Sin embargo, no es cardioselectiva y produce efectos neurológicos que impiden su uso en la clínica.
- AVE011863: bloqueador selectivo de ITO e IKur y sin efecto sobre IKr, por tanto normaliza el ECG y suprime la reentrada de fase 2 en el modelo de la porción de miocardio perfundida; sin embargo, tiene la desventaja de poseer efecto del primer paso en el hígado, por lo que no es efectiva oralmente.
- TEDISAMIL: Es un bloqueador de ITO que puede ser más potente que la quinidina por su escaso efecto en las corrientes de entrada; sin embargo, bloquea IKr y produce formas adquiridas de QT largo, por lo que puede cambiar un tipo de TVP por otro, además todavía está en evaluación22.
- ABRIDORES DE INa: Teóricamente sería un blanco ideal porque se opondría este
efecto a la corriente ITO disminuyendo
Otros fármacos estudiados62
- DISOPIRAMIDA: normaliza el ST en algunos pacientes con SB y lo desenmascara en otros.
- PROPAFENONA, FLECAINIDA y PROCAINAMIDA: contraindicados. Inducen el fenotipo del SB.
- AMIODARONA y ß-BLOQUEANTES: demostrada su ineficacia.
Queremos concluir esta profunda revisión recordando la idea con la que comenzamos nuestro artículo, sobre el necesario conocimiento de fisiología básica en este mundo de las arritmias. Katz afirmó en su clásico texto "Physiology of the heart", que las observaciones clínicas y los estudios controlados aleatorizados han dejado bien claro que los tratamientos carentes de una sólida base fisiológica pueden ocasionar más daño que beneficio70, ratificando esta expresión recordamos, al tal vez más reconocido clínico de todos los tiempos, Sir William Osler, quien afirmó: "un médico que no practique la fisiología es una especie de pistolero de farmacia disparándole a la enfermedad y al paciente, él mismo no sabría a cuál".
Referencias bibliográficas
1. Amin AS, Asghari-Roodsaria A, Tan HL. Cardiac sodium channelopathies. Pflugers Arch. 2009 Nov [Epub ahead of print].
2. Nerbonne JM, Kass RS. Molecular physiology of cardiac repolarization. Physiol Rev. 2005;85:1205-53.
3. Ter Keurs HEDJ, Boyden PA. Calcium and arrhythmogenesis. Physiol Rev. 2007; 87:457-06.
4. Antzelevitch C. Molecular genetics of arrhythmias and cardiovascular conditions associated with arrhythmias. Heart Rhythm. 2004;1(5):42-56.
5. BezzinaCR, Wilde AA, Roden DM. The molecular genetics of arrhythmias. Cardiovasc Res. 2005;67:343-6.
6. Antzelevitch C, Brugada P, Brugada J, Brugada R. The Brugada syndrome.From cell to bedside. Curr Probl Cardiol. 2005;30:9-54.
7. BezzinaCR, Shimizu W, Yang P. Common sodium channel promoter haplotype in Asian subjects underlies variability in cardiac conduction. Circulation. 2006;113: 338-44.
8. London B, Michalec M, Mehdi H. Mutation in glycerol-3-phosphate dehydrogenase 1 like gene (GPD1-L) decreases cardiac Na+ current and causes inheretid arrhythmias. Circulation. 2007;116:2260-68.
9. Antzelevitch C, Pollevick GD, Cordeiro JM, Casis O, Sanguinetti MC, Aizawa y, et al. Loss of function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation. 2007;115:442-9.
10. Antzelevitch C, Nof E. Brugada syndrome: Recent advances and controversies. Curr Cardiol Rep. 2008;10:376-83.
11. Delpon E, Cordeiro JM, Nuñez L, Thomsen PEB, Guerchicoff A, Pollevick GD, Wu y, et al. Functional effects of KCNE3 mutation and its role in the development of Brugada syndrome. Circ Arrhythmia Electrophysiol. 2008;1:209-18.
12. Massanet R, Gallardo-Chacon JJ, Caminal P, Perera A. Search of phenotype related candidate genes using gene ontology-based semantic similarity and protein interaction information: Aplication to Brugada syndrome. Conf Proc IEEE Eng Med Biol Soc. 2009;1:7079-82.
13. Antzelevitch C, Dumaine R. Electrical heterogeneity in the heart: Physiological, pharmacological and clinical implications. In: Page E, Fozzard HA, Solaro RJ, eds. Handbook of physiology. Section 2. The cardiovascular system. New York:Oxford University Press. 2001:654-92.
14. Antzelevitch C. Cellular basis and mechanism underlying normal and abnormal myocardial repolarization and arrhythmogenesis. Ann Med. 2004;36(suppl 1):5-14.
15. Antzelevitch C, Viskin S. Brugada syndrome: cellular mechanisms and approaches tp therapy. In: Gussak I, Antzelevitch C, eds. Electrical diseases of the heart. Genetics, mechanisms, treatment,prevention. Part III. Chapter 33: Spinger London; 2008. p. 500-35.
16. Benito B, Brugada R, Brugada J, Brugada P. Brugada syndrome. Prog Cardiovasc Dis. 2008;51(1):1-22.
17. Ohkubok, Watanabe I, Okumura Y, Takagi Y, Ashino S, Kofune M, et al. Right ventricular histological substrate and conduction delay in patients with Brugada syndrome. Int Heart J. 2010;51(1):17-23.
18. Toh N, Morita H, Nagase S, Tanigushi M, Miura N, Nishii N, et al. Atrial electrophysiological and structural remodeling in high-risk patients with Brugada syndrome: assessment with electrophysiology and echocardiography. Heart Rhythm. 2010;7(2):218-24.
19. Postema PG, van Dessel PF, Kors JA, Linnenbank AC, van Herpen G, Ritsema van Eck HJ, et al. Local depolarization abnormalities are the dominant pathophysiologic mechanism for type 1 electrocardiogram in Brugada syndrome a study of electrocardiograms, vectorcardiograms, and body surface potential maps during ajmaline provocation. J Am Coll Cardiol. 2010;55(8):789-97.
20. Antzelevitch C. In vivo human demostration of phase 2 reentry. Heart Rhythm. 2005;2(8):804-06.
21. Thomsen PEB, Joergensen RM, Kanters JK, Jensen JT, Haarbo J, Hagemann A, et al. Phase 2 reentry in man. Heart Rhythm. 2005;2:797-03.
22. Antzelevitch C. Brugada syndrome. PACE. 2006;29:1130-59.
23. Fish JM, Antzelevitch C. Link between hypothermia and the Brugada syndrome. J Cardiovasc Electrophysiol. 2004;15:942-44.
24. Nishida K, Fujuki A, Mizumaki K, Sakabe M, Sugao M, Tsuneda T, et al. Canine model of Brugada syndrome using regional epicardial cooling of the right ventricular outflow tract. J Cardiovasc Electrophysiol. 2004;15:936-41.
25. Antzelevitch C, Brugada P, Borggrefe M, Brugada J, Brugada R, Corrado D, et al. Brugada syndrome. Report of the second consensus conference. Circulation. 2005;111:659-70.
26. Antzelevitch C, Brugada R. Fever and the Brugada syndrome. PACE. 2002;25: 1637-39.
27. Ambardekar AV, Lewkowiez L, Krantz MJ. Mastitis unmasks Brugada syndrome. Int J Cardiol. 2009;132(3):e94-6.
28. Makaryus JN, Verbsky J, Schwartz S, Slotwiner D. Fever associated with gastrointestinal shigellosis unmasks probable Brugada syndrome. Case Report Med. 2009;2009:492031.
29. Samani K, Wu G, Ai T, Shuraih M, Mathuria NS, Li Z, et al. A novel SCN5A mutation V1340I in Brugada syndrome augmenting arrhythmias during febrile illness. Heart Rhythm. 2009;6(9):1318-26.
30. Amin AS, Klemens CA, Verkerk AO, Meregalli PG, Asghari-Roodsari A, de Baker JM. Fever-triggered ventricular arrhythmias in Brugada syndrome and type 2 long-QT. Neth Heart J. 2010;18(3):165-9.
31. Yamaki M, Sato N, Myojo T, Nishiura T, Nishimura M, Nakamura H, et al. Possible contribution of ischemia of the conus branch to induction or augmentation of Brugada type electrocardiographic changes in patients with coronary artery disease. Int Heart J. 2010;51(1):68-71.
32. Hu D, Viskin S, Oliva A, Carrier T, Cordeiro JM, Barajas-Martinez H, et al. Novel mutation in the SCN5A gene associated with arrythmic storm development durin acute myocardial infarction. Heart Rhythm. 2007;4:1072-80.
33. Hu D, Viskin S, Oliva A, Cordeiro JM, Guerchicoff A, Pollevick GD, et al. Genetic predisposition and cellular basis for ischemia-induced ST-segment changes and arrhythmias. J Electrocardiol. 2007;40:526-9.
34. Cordeiro JM, Mazza M, Goodrow R, Ulahannan N, Antzelevitch C, DiDiego JM. Functionally distinct sodium channels in ventricular epicardial and endocardial cells contribute to a greater sensitivity of the epicardium to electrical depression. Am J Physiol Heart Circ Physiol. 2008;295:H154-H62.
35. Jorens PG, Van den Eynden GG. Propofol infusion síndrome with arrhythmia myocardial fat accumulation and cardiac failure. Am J Cardiol. 2009;104(8):1160-2.
36. Vernooy K, Delhaast T, Cremer OL, DiDiego JM, Oliva A, Timmermans C, et al. Electrocardiographic changes predicting sudden death in propofol-related infusion syndrome. Heart Rhythm. 2006;3:131-37.
37. Barajas-Martínez HM, Hu D, Cordeiro JM, Wu Y, Kovacs RJ, Meltser H, et al. Lidocaine-induced Brugada syndrome phenotype linked to a novel mutation in the cardiac sodium channel. Circ Res. 2008;103:396-04.
38. Chandra PA, Chandra AB. Brugada syndrome unmasked by lithium. South Med J. 2009;102(12):1263-5.
39. Meert A, Vermeersch N, Beckers R, Hoste W, Brugada P, Hubloue I. Brugada-like ECG pattern induced by tricyclic antidepressants. Eur J Emerg Med. 2009 Dec 5. [Epub ahead of print].
40. Chen PS, Priori SG. The Brugada syndrome. J Am Coll Cardiol. 2008;51(12): 1176-80.
41. Kusano KF, Taniyama M, Nakamura K, Miura D, Banba K, Nagase S, et al. Atrial fibrillation in patients with Brugada syndrome. J Am Coll Cardiol. 2008;51(12): 1169-75.
42. Francis J, Antzelevitch C. Atrial fibrillation and Brugada syndrome. J Am Coll Cardiol. 2008;51(12):1149-53.
43. Kostopoulou A, Koutelou M, Theodorakis G, Theodorakos A, Livanis E, et al. Disorders of the autonomic nervous system in patients with Brugada syndrome: a pilot study. J Cardiovasc Electrophysiol. 2010 Feb 1. [Epub ahead of print].
44. Brugada P, Benito B, Brugada R, Brugada J. Brugada syndrome: up date 2009. Hellenic J Cardiol. 2009;50(5):352-72.
45. Wolpert C, Echternach C, Veltmann C. Intravenous drug challenge using flecainide and ajmaline in patients with Brugada syndrome. Heart Rhythm. 2005;2:254-60.
46. Sarkozy A, Chierchia GB, Paparella G, Boussy T, De Asmundis C, Roos M, et al. Inferior and lateral electrocardiographic repolarization abnormalities in Brugada syndrome. Circ Arrhythm Electrophysiol. 2009;2(2):154-61.
47. Miyamoto K, Yokokawa M, Tanaka K. Diagnostic and prognostic value of a type 1 Brugada electrocardiogram at higher (third or second) V1 to V2 recording in men with Brugada syndrome. Am J Cardiol. 2007;99:53-7.
48. Batchvarov VN, Govindan M, Macfarlane P, Camm AJ, Behr ER. Diagnostic utility of bipolar precordial leads during ajmaline testing for suspected Brugada syndrome. Heart Rhythm. 2010;7(2):208-15.
49. Carlsson J, Erdogan A, Shutle B, Neuzner J, Pitchner HF. Possible role of epicardial left ventricular programmed stimulation in Brugada syndrome. Pacing Clin Electrophysiol. 2001;24:247-9.
50. Paul M, Gerss J, Shulzc-Bahr E, Wichter T, Vahlhaus C, Wilde AAM, et al. Role of programmed ventricular stimulation in patients with Brugada syndrome: a metaanalysis of worldwide published data. Eur Heart J. 2007;28(17):2126-33.
51. Viskin S, Rogowski O. Asymptomatic Brugada syndrome: a cardiac ticking time-bomb? Europace. 2007;9(9):707-10.
52. Priori SG, Napolitano C. Management of patient with Brugada syndrome should not be based on programmed electrical stimulation. Circulation. 2005;111:285-91.
53. Eckardt L, Probst V, Smits JP. Long-term prognosis of individuals with right precordial ST-segment-elevation Brugada syndrome. Circulation. 2005;111:257-63.
54. Probst V, Veltmann C, Eckardt L, Meregall PG, Gaita F, Tan HL, et al. Long.-term prognosis of patients diagnosed with Brugada syndrome. Results from the FINGER Brugada syndrome registry. Circulation. 2010 Jan 25[Epub ahead of print].
55. Viskin S, Wilde AAM, Tan HL, Antzelevitch C, Shimizu W, Belhassen B. Empiric quinidine therapy for asymptomatic Brugada syndrome: Time for a prospective registry. Heart Rhythm. 2009;6:401-4.
56. Kamakura S, Ohe T, Nakazawa K, Aizawa Y, Shimizu A, Horie M, et al. Long.term prognosis of probands with Brugada-pattern ST-elevation in leads V1-V3. Circ Arrhythm Electrophysiol. 2009;2(5):495-03.
57. Nakano Y, Shimuzu W, Ogi H, Suenari K, Oda N, Makita Y, et al. A spontaneous type 1 electrocardiogram pattern in lead V2 is an independent predictor of ventricular fibrillation in Brugada syndrome. Europace. 2010 Jan 31. [Epub ahead of print].
58. Meregalli PG, Ruijter JM, Hofman N. Diagnostic value of flecainide testing in unmasking SCN5A-related Brugada syndrome. J Cardiovasc Electrophysiol. 2006;17:857-64.
59. Fish JM, Antzelevitch C. Cellular mechanism and arrhythmogenic potential of T-wave alternans in the Brugada syndrome. J Cardiovasc Electrophysiol. 2008;19: 301-08.
60. Das MK, El Masry H. Fragmented QRS and other depolarization abnormalities as a predictor of mortality and sudden cardiac death. Curr Opin Cardiol. 2010; 25(1):59-64.
61. Hayashi H, Sumiyoshi M, Yasuda M, Komatsu K, Sekita G, Kawano Y. Prevalence of the Brugada-type electrocardiogram and incidence of Brugada syndrome in patients with sick sinus syndrome. Circ J. 2010;74(2):271-7.
62. Ikeda T, Abe A, Yusu S, Nakamura K, Ishiguro H, Mera H, et al. The full stomach test as a novel diagnostic technique for identifying patients at risk of Brugada syndrome. J Cardiovsc Electrophysiol. 2006;17:602-07.
63. Antzelevitch C, Fish JM. Therapy for the Brugada syndrome. In: Kass RS, Clancy CE, eds. Handbook of experimental pharmacology. Vol. 171. Basis and treatment of cardiac arrhythmias. New York: Springer; 2006.p. 305-30.
64. Veltmann C, Kuschuk J, Schimp FR, Streitner F, Shoene N, Borggrefe M, et al. Prevention of inappropriate ICD shocks in patients with Brugada syndrome. Clin Res Cardiol. 2010;99(1):37-44.
65. Carmona RP, Ramos RR, Padrón GP, Rodríguez AL. Nuevas indicaciones de la quinidina la rescatan del olvido. Rev Clin Esp. 2008;208(7):369-73.
66. Yang F, Hanon S, Lam P, Shweitzer P. Quinidine revisited. Am J Med. 2009; 122(4):317-21.
67. Carmona RP, Ramos RR, Padrón GP, Chávez EG, Rodríguez AL, Cruz JME, et al. La nueva era de la quinidina amenazada de extinción. Rev Fed Arg Cardiol. 2009;38(3):173-5.
68. Shine HC, Divakaran VG, Lakkis NM. A case of Brugada syndrome presenting with incessant polymorphic ventricular tachycardia. Clin Cardiol. 2010 Feb 2. [Epub ahead of print].
69. Fish JM, Welchons DR, Kim YS, Lee SH, Ho WK, Antzelevitch C. Dimetyl lithoespermate B, an extract of Danshen, suppresses arrhythmogenesis associated with the Brugada syndrome. Circulation. 2006;113(11):1393-400.
70. Katz AM, editors. Preface. En: Physiology of the heart. 4ta ed. Philadelphia: Lipincott Williams & Wilkins; 2006. p. 1.
Recibido: 10 de mayo de 2010
Aceptado para su publicación: 31 de mayo de 2010